[CANCER RESEARCH 47, 1199-1220, March 1, 1987]
Perspectives in Cancer Research
Retroviruses as Carcinogens and Pathogens: Expectations and Reality
Peter H. Duesberg
Department of Molecular Biology and Virus Laboratory, University of California, Berkeley, California 94720
Abstract
Retroviruses (without transforming genes) are thought to cause leukemia's and other cancers in animals and humans because they were originally isolated from those diseases and because experimental infections of new-borns may induce leukemia's with probabilities of 0 to 90%. According to this hypothesis viral cancers should be contagious, polyclonal, and preventable by immunization. However, retroviruses are rather widespread in healthy animals and humans where they typically cause latent infections and antiviral immunity. The leukemia risk of such infections is less than 0.1% and thus about as low as that of virus-free controls. Indeed retroviruses are not sufficient to initiate transformation (a) because of the low percentage of symptomatic virus carriers and the complete lack of transforming function in vitro; (b) because of the striking discrepancies between the long latent periods of 0.5 to 10 years for carcinogenesis and the short eclipse of days to weeks for virus replication and direct pathogenic and immunogenic effects; © because there is no gene with a late transforming function, since all genes are essential for replication; (d) because host genes, which do not inhibit virus, inhibit tumorigenesis up to 100% if intact and determine the nature of the tumor if defective; and above all (e) because of the monoclonal origin of viral leukemias, defined by viral integration sites that are different in each tumor. On these bases the probability that a virus-infected cell will become transformed is estimated to be about 10 (11th power). The viruses are also not necessary to maintain transformation, since many animal and all bovine and human tumors do not express viral antigens or RNA or contain only incomplete proviruses. Thus as carcinogens retroviruses do not or only very rarely (10 (11th power)) fulfill the third. Therefore it has been proposed that retroviruses transform inefficiently by activating latent cellular oncogenes by for example provirus integration. This predicts diploid tumors with great diversity, because integration sites are different in each tumor. However, the uniformity of different viral and even nonviral tumors of the same lineage, their common susceptibility to the same tumor resistance genes, and transformation-specific chromosome abnormalities shared with nonviral tumors each argue for cellular transforming genes. Indeed clonal chromosome abnormalities are the only known transformation-specific determinants of viral tumors. Since tumors originate with these abnormalities, these or associated events, rather than preexisting viruses, must initiate transformation. Therefore it is proposed that transformation is a virus-independent event and that clonal viral integration sites are consequences of clonal proliferation of transformed cells. The role of the virus in carcinogenesis is limited to the induction of hyperplasia which is necessary but not sufficient for carcinogenesis. Hyperplasia depends on chronic viremia or high virus expression which are very rare in animals outside the laboratory and have never been observed in humans. Since latent viruses, which are typical of nearly all natural infections, are neither direct nor indirect carcinogens, they are not targets for cancer prevention. Viruses are also not targets for cancer therapy, since tumors are not maintained and not directly initiated by viral genes and occur naturally despite active antiviral immunity.
Lymphotropic retrovirus has been proposed to cause AIDS because 90% of the patients have antibody to the virus. Therefore antibody to the virus is used to diagnose AIDS and those at risk for AIDS. The virus has also been suggested as a cause of diseases of the lung and the nervous system. Promiscuous male homosexuals and recipients of frequent transfusions are at high risk for infection and also at a relatively high risk for AIDS, which averages 0.3% and may reach 5%. Others are at a low risk for infection and if infected are at no risk for AIDS. AIDS viruses are thought to kill T-cells, although these viruses depend on mitosis for replication and do not lyse cells in asymptomatic infections. Indeed the virus is not sufficient to cause AIDS (a) because the percentage of symptomatic carriers is low and varies between 0 and 5% with the risk group of the carrier, suggesting a cofactor or another cause; (b) because the latent period for AIDS is 5 years compared to an eclipse of only days to weeks for replication and direct pathogenic and immunogenic effects; and © because there is no gene with a late AIDS function, since all viral genes are essential for replication. Moreover the extremely low levels of virus expression and infiltration cast doubt on whether the virus is even necessary to cause AIDS or any of the other diseases with which it is associated. Typically, proviral DNA is detectable in only 15% of AIDS patients and then only in one of 10 (2nd power) to 10 (3rd power) lymphocytes and is expressed in only 1 of 10 (4th power) to 10 (5th power) lymphocytes. Thus the virus is inactive or latent in carriers with and without AIDS. It is for this reason that it is not transmitted as a cell-free agent. By contrast, all other viruses are expressed at high titers when they function as pathogens. Therefore AIDS virus could be just the most common occupational infection of those at risk for AIDS because retroviruses are not cytocidal and unlike most viruses persist as latent, nonpathogenic infections. As such the virus is an indicator of sera that may cause AIDS. Vaccination is not likely to benefit virus carriers, because nearly all have active antiviral immunity.
How often have I said to you, that when you have eliminated the impossible, whatever remains however improbable must be the truth.
-- Sherlock Holmes
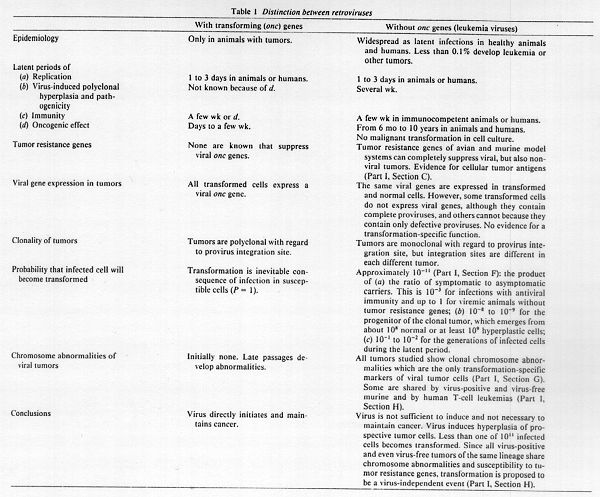
The irreversible and predictable courses of most cancers indicate that cancer has a genetic basis. In 1914 Boveri (1) proposed that cancer is caused by chromosomal mutations. This hypothesis has since received ample support (2-4), although a cellular cancer gene has yet to be identified (5). In the light of the spectacular discovery of RSV* in 1911, which proved to be a direct, infectious carcinogen, the hypothesis emerged that viruses may be a significant source of exogenous cancer genes (6). The virus-cancer hypothesis has since steadily gained support because retroviruses and DNA viruses were frequently isolated from animal leukemias and other tumors, and occasionally from human leukemias, in efforts to identify causative agents (7-16). However, once discovered in tumors and named tumor viruses, most of these viruses were subsequently found to be widespread in healthy animals and humans (8, 12-18). Thus these viruses are compatible with the first but apparently not necessarily with the third of Koch's postulates** as viral carcinogens. Only a few of the many tumor viruses are indeed directly oncogenic, such as RSV and about 20 other types of retroviruses (5, 13, 19, 20), and hence compatible with Koch's third postulate. Therefore, if we want to assess the role of viruses in cancer, there must be a clear separation between those viruses which are directly oncogenic and those which are not. The directly oncogenic retroviruses owe their transforming function to a particular class of genes which are termed onc genes (20). These are as yet the only known autonomous cancer genes that can transform diploid cells in vitro as well as in animals susceptible to the particular virus (5). Since susceptible cells are inevitably transformed as soon as they are infected, the resulting tumors are polyclonal (13, 16). Nevertheless, directly oncogenic retroviruses have never caused epidemics of cancer. The probable reason is that onc genes are not essential for the survival of the virus and hence are readily lost by spontaneous deletion or mutation (5). Indeed, onc genes were originally discovered by the analysis of spontaneous onc deletion mutants of RSV (21). Moreover, because onc genes typically replace essential genes (except in some strains of RSV) these viruses cannot replicate unless aided by a nondefective helper virus (5, 13).
The vast majority of the tumor viruses are retroviruses and DNA viruses that do not contain onc genes. The RNA genomes of all retroviruses without onc genes measure only 8 or 9 kilobases (13, 22). They all encode three major essential genes which virtually exhaust their coding capacity. These are in the 5' to 3' map order gag which encodes the viral core protein, pol which encodes the reverse transcriptase, and env which encodes the envelope glycoprotein (23, 24). Although these viruses lack onc genes they are considered tumor viruses, because they were originally isolated from tumors and because experimental infections may induce tumors under certain conditions. However, in contrast to tumors caused by viruses with onc genes, such tumors are always monoclonal and induced reproducively only in genetically selected animals inoculated as newborns after latent periods of over 6 months (see below). Because of the long latent periods, these retroviruses are said to be "slow" viruses (13, 16), although their mechanism of replication is exactly the same as that of their fast and efficient relatives with onc genes that transform cells as soon as they infect them (5, 19) (Table 1). The retroviruses are also considered to be plausible natural carcinogens because they are not cytocidal and hence compatible with neoplastic growth and other slow diseases. Indeed, retroviruses are the only viruses that depend on mitosis for replication (13, 25).
However, the retroviruses without onc genes are also the most common and benign passenger viruses of healthy animals and humans probably because of their unique noncytocidal mechanism of replication and their characteristic ability to coexist with their hosts without causing any pathogenic symptoms either as latent infections, which make no biochemical demands, or even as productive infections. Based on the permissiveness of a host for expression and reproduction, they have been divided into exogenous viruses which are typically expressed and hence potentially pathogenic and endogenous viruses which are typically latent and hence nonpathogenic (16-18). Because they are so readily suppressed in response to as yet undefined cellular suppressors (8, 11, 12, 16-18), endogenous viruses are integrated as proviruses into the germ line of most if not all vertebrates (8, 13, 16-18). Nevertheless, the endogenous and exogenous retroviruses are entirely isogenic and there is no absolute biochemical or functional distinction between them except for their response to suppressors of a particular host (13, 16-18) (Part I, Section A). Therefore the association of these viruses with a given disease is not sufficient even to suggest a causative role in it. Indeed there is as yet no direct evidence that retroviruses play a role as natural carcinogens of wild animals and humans. Thus the critical expectations of the virus-cancer hypothesis, namely that RNA or DNA tumor viruses would be direct carcinogens, that viral tumors would be polyclonal because each virus-infected cell would be transformed, and above all that viral carcinogenesis would be preventable by immunization, remain largely unconfirmed.
Recently retroviruses without onc genes have been isolated from patients with AIDS and those at risk for AIDS and have since been considered the cause of AIDS (26). In contrast to other retroviruses, the AIDS viruses are thought to act as direct, cytocidal pathogens that kill susceptible T-cells (13, 27).
Here we discuss how the retroviruses without onc genes fit the role of viral carcinogens or AIDS pathogens and whether these viruses are indeed the vessels of evil they have been labeled to be. Above all we hope to identify transformation-specific or AIDS-specific viral and cellular determinants and functions. Since the genetic repertoire of all retroviruses without onc genes, including that of the AIDS viruses (28), is exhausted by genes that are essential for virus replication (13,24), a hypothetical oncogenic or AIDS function would have to be indirect or it would have to be encoded by one of the essential genes. In the second case the virus would be oncogenic or cause AIDS wherever it replicates. A survey of the best studied animal and human retroviruses demonstrates that these viruses are not sufficient to cause tumors and not necessary to maintain them. Most likely these viruses play a role in inducing tumors indirectly. Indeed transformation appears to be a virus-independent, cellular event for which chromosome abnormalities are the only specific markers. Likewise the AIDS viruses are shown not to be sufficient to cause AIDS, and the evidence that they are necessary to cause it is debated.
1. Retroviruses and Cancer
A. Retroviruses Are Not Sufficient for Transformation Because Less Than 0.1% of Infected Animals or Humans Develop Tumors
Avian lymphomatosis virus was originally isolated from leukemic chickens (29). However, subsequent studies proved that latent infection by avian lymphomatosis viruses occurs in all chicken flocks and that by sexual maturity most birds are infected (30-32). Statistics report an annual incidence of 2 to 3% lymphomatoses in some inbred flocks. Yet these statistics include the more common lymphomas caused by Marek's virus (a herpes virus) (33, 34). The apparent paradox that the same virus is present in most normal and healthy animals (30) but may be leukemogenic in certain conditions was resolved at least in descriptive terms by experimental and congenital contact infections. Typically experimental or contact infection of newborn animals that are not protected by maternal antibody would induce chronic (31, 32) or temporal (35, 36) viremia. The probability of such animals for subsequent lymphomatosis ranges from 0 to 90% depending on tumor resistance genes (Section C). However, infection of immunocompetent adults or of newborn animals protected by maternal antibody and later by active immunity would induce latent, persistent infections with a very low risk of less than 1% for lymphomatosis (32, H. Rubin, personal communication.) Thus only viremic animals are likely to develop leukemia at a predictable risk.
Viremia has a fast proliferative effect on hemopoietic cells and generates lymphoblast hyperplasia (Fig. 1) (32, 36, 37). Hyperplasia appears to be necessary but not sufficient for later leukemogenesis because it does not lead to leukemia in tumor-resistant birds (36) (Section C) and because removal of the burso of Fabricius, the major site of lymphoproliferation, prevents development of the disease (9, 32).
The murine leukemia viruses were also originally isolated from leukemic inbred mice (9) and subsequently detected as latent infections in most healthy mice (8, 13, 16, 17, 38). Indeed, about 0.5% of the DNA of a normal mouse is estimated to be proviral DNA of endogenous retroviruses, corresponding to 500 proviral equivalents per cell (18). Nevertheless leukemia in feral mice is apparently very rare. For instance low virus expression, but not a single leukemia, was recorded in 20% of wild mice (38) probably because wild mice restrict virus expression and thus never become viremic and leukemic. However, in an inbred stock of feral mice predisposed to lymphoma and paralysis, 90% were viremic from an early age, of which 5% developed lymphomas at about 18 months (39).
Experimental infections of newborn, inbred mice with appropriate strains of murine leukemia viruses induce chronic viremias. Such viremic mice develop leukemias with probabilities of 0 to 90% depending on the mouse strain (Section C). However, if mice that are susceptible to leukemogenesis are infected by the time they are immunocompetent or are protected by maternal antibodies if infected as neonates, no chronic viremia and essentially no leukemia are observed (although a latent infection is established) (41). Thus leukemogenesis depends on viremia (40) as with the avian system. However, viremia is not sufficient, because certain tumor-resistant strains do not develop leukemia even in the presence of viremia (42) (Section C). Again viremia has an early proliferative effect on lymphocytes which has been exploited to quantitate these viruses in vivo within 2 weeks by the "spleen weight" or "spleen colony" assay (18, 43-47). This hyperplasia of lymphocytes is necessary for leukemogenesis, because the risk that an infected animal will develop leukemia is drastically reduced or eliminated by thymectomy, which is a major source of cells for prospective leukemogenesis (9).
The AKR mouse is a special example in which spontaneous expression of endogenus virus and the absence of tumor resistance genes inevitably lead to viremia at a few weeks after birth and, in 90% of the animals, to leukemia at 6 to 12 months of age (9, 41, 48). This also shows that endogenous viruses can be just as pathogenic or leukemogenic as exogenous viruses if they are expressed at a high level. Likewise, endogenous avian retroviruses are leukemogenic in chickens permissive for acute infection (49, 50).
The evidence that mammary carcinomas are transmissible by a milk-borne virus, MMTV, indicates that the virus is an etiological faction (51, 52). However, the same virus is also endogenous but not expressed in most healthy mice (16, 53). Since no mammary tumors have been reported in wild mice the natural incidence must be very low, but in mice bred for high incidence of mammary carcinomas it may rise to 90% (13, 16, 54, 55). As with the leukemia viruses, the risk for tumorigenesis was shown to depend on a high level of virus expression from an early age and on the development of hyperplasias that are necessary but not sufficient for carcinogenesis (56, 57). For example, BALB/c mice that express over 100 mu-g virus per ml milk all develop tumors after latencies of over 12 months, but mice that express 3 mu-g or less virus per ml develop no tumors at all (54, 58).
Feline leukemia virus was originally isolated from cats with lymphosarcoma (59) and subsequently from many healthy cats. It is estimated that at least 50 to 60% of all cats become naturally infected by feline leukemia viruses at some time during their lives (60, 61). However, only about 0.04% of all cats develop leukemia on an annual basis (62), which is thought to be caused by these viruses (13, 61, 63). Most natural infections cause transient virus expression which is followed by an immune response, after which little virus is expressed (60, 64, 65). Such infections do not induce leukemias at a predictable rate (61). However, 1 to 2% of the naturally infected cats become chronically viremic (66). About 28% of the viremic cats develop leukemias after latent periods of 2 years. Thus viremia indicates a high risk for the development of leukemia (66). Viremia may result from a congenital infection in the absence of maternal antibody or from a native immunodeficiency. As in the avian and murine systems, experimental infection of newborn, immunotolerant cats produces early viremia and runting diseases and late leukemias at a much higher incidence than natural infections (63, 64, 67, 68). The gibbon ape leukemia virus was also initially discovered in leukemic apes and was later isolated from healthy gibbons (13, 69). Again, only chronically viremic gibbons were shown to be at risk for leukemia (70).
The bovine and human retroviruses associated with acute leukemias are always biochemically inactive or latent (Section D). Viremia, which is frequently associated with a leukemia of congenitally or experimentally infected domestic chickens, cats, or inbred mice, has never been observed in the bovine or human system. Accordingly bovine and human leukemia viruses could be isolated from certain leukemic cells only after cultivation in vitro away from the suppressive immune system of the host (71, 72). In regions of endemic bovine leukemia virus infection 60 to 100% of all animals in a herd were found to contain antiviral antibody (73, 74). However, the incidence of leukemia was reported to range only from 0.01 to 0.4% (16, 73). Experimental infections with cell-free virus have not provided conclusive evidence for viral leukemogenesis. As yet only 1 of 25 animals infected with bovine leukemia virus has developed a leukemia 7 years after inoculation (73). Additional inoculations of 20 newborn calves did not cause a single leukemia within 5 years, although all animals developed antiviral antibody. [J. M. Miller and M. S. Van der Maaten, personal communication.] However, 50% of newborn sheep inoculated with bovine leukemia virus developed leukemia about 4 years later (75). These sheep were probably more susceptible to the bovine virus than cattle, because they would lack maternal antibody to the virus. Indeed they could have been transiently viremic, because antibody was detected only 4 months after inoculation (75).
HTLV-1 or ATLV was originally isolated from a human cell line derived from a patient with T-cell leukemia (71). It replicates in T-cells (27) and also in endothelial cells (76) or fibroblasts (77). The virus was subsequently shown, using antiviral antibody for detection, to be endemic as latent, asymptomatic infections in Japan and the Caribbean (27). Since virus expression is undetectably low not only in healthy but also in leukemic virus carriers, infections must be diagnosed indirectly by antiviral antibody or biochemically by searching for latent proviral DNA (Section D). Due to the complete and consistent latency, the virus can be isolated from infected cells only after activation in vitro when it is no longer controlled by the host's antiviral immunity and suppressors. Therefore the virus is not naturally transmitted as a cell-free agent like other pathogenic viruses, but only congenitally, sexually, or by blood transfusion, that is, by contacts that involve exchange of infected cells (13, 27).
It is often pointed out that functional evidence for the virus-cancer hypothesis is difficult to obtain in humans because experimental infection is not possible and thus Koch's third postulate cannot be tested. However, this argument does not apply here since naturally and chronically infected, asymptomatic human carriers are abundant. Yet most infections never lead to leukemias and none have ever been observed to cause viremias. Moreover, not a single adult T-cell leukemia was observed in recipients of blood transfusions from virus-positive donors (13, 78, 79), although recipients developed antiviral antibody (81).
The incidence of adult T-cell leukemia among Japanese with antiviral immunity is estimated to be only 0.06% based on 339 cases of T-cell leukemia among 600,000 antibody-positive subjects (78). Other studies have detected antiviral antibody in healthy Swedish donors (268) and in 3.4% of 1.2 x 10 (6 power) healthy Japanese blood donors (79). Further, it was reported that 0.9% of the people of Taiwan are antibody positive, but the incidence of the leukemia was not mentioned (80).
In conclusion, the tumor risk of the statistically most relevant group of retrovirus infections, namely the latent natural infections with antiviral immunity, is very low. It averages less than 0.1% in different species, as it is less than 1% in domestic chickens, undetectably low in wild mice, 0.04% in cattle, and 0.06% in humans. Thus the virus is not sufficient to cause cancer.
Moreover, since the viruses associated with all human tumors and most natural tumors of animals are latent and frequently defective (Section D), it is difficult to justify the claims that these viruses play any causative role in tumorigenesis. Indeed nearly all healthy chickens, mice, cats, cattle, and humans carry endogenous and exogenous retroviruses that are latent and hence neither pathogenic nor oncogenic (12, 16-18, 78, 82). Latent infections by cytocidal viruses, such as herpes viruses, are likewise all asymptomatic (83). Nevertheless it may be argued that only a small percentage of retroviral infections are expected to be oncogenic because only a small percentage of all other viral or microbial infections are pathogenic. However, the low percentage of symptomatic infections with other viruses and microbes reflects the low percentage of acute infections that have overwhelmed host defense mechanisms, but not a low percentage of latent infections that cause disease. Thus there is no orthodox explanation for the claims that some murine and avian, most feline, and all bovine and human leukemias (Section D) are the work of latent viruses.
Even the view that retroviruses cause leukemia or carcinoma directly in productive infections is debatable, because indeed highly productive infections are frequently asymptomatic. For example, despite chronic acute viremias certain chickens, mice, or cats, inoculated experimentally or by contact as immuno-tolerant newborns, do not develop leukemia (see above and Section C). Further no malignant transformation has ever been observed in cultured cells that are actively producing retroviruses, and the probability that an infected cell of an animal will become transformed is only 10 (11th power) (Section F). This low probability that a productively infected cell will become transformed is a uniquely retrovirus-specific reason for asymptomatic infections. It is for this reason that retroviruses without onc gene can be asymptomatic for cancer even in acute, productive infections of animals (30, 31, 36, 42, 66, 70), although they may then cause other diseases (Section B).
Thus retrovirus infections are not only asymptomatic due to latency and low levels of virus infiltration, like all other viruses, but are also asymptomatic due to a particular discrepancy between acute and productive infection and oncogenesis. To answer the question of why some viremic animals do and others do not develop leukemia and why tumors appear so late after infection (Section B), both tumor resistance genes (Section C) and the mechanism of transformation must be considered (Section H).
B. Discrepancies between the Short Latent Period of Replication and the Long Latent Periods of Oncogenesis: Further Proof That Virus Is Not Sufficient for Cancer
Here we compare the kinetics of virus replication and direct pathogenic and immunogenic effects with the kinetics of virus-induced transformation. If retroviral genes were sufficient to induce cancer, the kinetics of carcinogenesis would closely follow the kinetics of virus replication.
Kinetics of Replication and of Early Pathogenic and Immunologenic Effects. The eclipse period of retrovirus replication has been determined to be 1 to 3 days in tissue culture (Table 1) using either transforming onc genes as markers or the appearance of reverse transcriptase or interference with other viruses or plaque formation for viruses without onc genes (13, 16) (see below). The incubation period following which retroviruses without onc genes induce viremia in animals is 1 to several weeks (9, 13, 14, 16) (Table 1). In immunocompetent animals antiviral immunity follows infections with a lag of 2 to 8 weeks.
In animals, retroviruses without onc genes can be directly pathogenic if they are expressed at high titers. For instance, avian retroviruses may cause in newborn chickens diseases of polyclonal proliferative nature like osteopetrosis, angiosarcoma, hyperthyroidism (84-87), or hyperplastic follicles of B-cells in the bursa of Fabricius (36, 37) after latencies of 2 to 8 weeks. The same viruses may also cause diseases of debilitative nature such as stunting, obesity, anemia, or immunodeficiency after lag periods of 2 to 8 weeks (88, 89). Infections of newborn mice that cause viremia also cause polyclonal lymphocyte hyperplasias, splenomegaly, and immuno-suppression several weeks after infection (47) (Section A). The early appearance of hyperplastic nodules in mammary tumor virus-infected animals prior to malignant transformation has also been proposed to be a virus-induced, hyperplastic effect (56, 57). Infection of newborn kittens with feline leukemia virus causes early runting effects and depletion of lymphocytes within 8 to 12 weeks (64, 67, 68) followed by persistent viremia in up to 80% of the animals (90). In experimentally infected adult animals mostly transient (85%) and only a few persistent (15%) viremias are observed (64, 68, 90). Likewise primate retroviruses such as Mason-Pfizer virus (91) or simian AIDS virus (92) or STLV-III virus (93) may cause runting, immuno-depression, and mortality several weeks after inoculation if the animals do not develop antiviral immunity. These early and direct pathogenic effects of retroviruses without onc genes depend entirely on acute infections at high virus titers and occur only in the absence of or prior to antiviral immunity.
Retroviruses have also been observed to be directly pathogenic by mutagenesis via provirus integration of cellular genes (13, 16, 94, 95). Given about 10 (6th power) kilobases for the eukaryotic genome and assuming random integration, a given cellular gene would be mutated in 1 of 10 (6th power) infected cells (see Sections E and F). Therefore this mechanism of pathogenesis would play a role in vivo only if mutagenesis were to occur at a single or few cell stage of development (94) or if such a mutation would induce clonal proliferation, as is speculated in Section E.
Certain direct, cytopathic effects of retroviruses without onc genes are also detectable in vitro within days or weeks after infection, although malignant transformation has never been observed in cell culture. For example, the avian reticulo-endotheliosis viruses fuse and kill a fraction of infected cells during the initial phase of infection (96, 97). Certain strains of avian retroviruses form plaques of dead primary chicken embryo cells in culture within 7 to 12 days postinfection. This effect is probably based on cell fusion and has been used as a reliable virus assay (45, 98). The plaque assays of murine leukemia viruses on XC rat cells (99) and on mink cells (101-104) also reflect fast cytopathic effects involving fusions of infected cells (45). Cell fusion of human lymphocytes in vitro is also typical of HTLV-I (105, 106) and of AIDS virus (27) (see Part II). Cells are thought to be fused in vitro by cross-linking through multivalent bonds between viral envelope antigens and cellular receptors, a process that requires high local concentration of virus particles (13, 16, 27, 45, 105). The fusion effect is not observed in chronic acute or latent infections of animals or humans or in chronically infected cell lines cultured in vitro. Therefore it appears to be predominantly a cell culture artifact, possibly resulting from interaction between virus receptors of uninfected cells with viruses budding from the surface of adjacent cells. This has been directly demonstrated by inhibition of HTLV-I-mediated fusion with antiserum from infected individuals (105). Thus as direct pathogens the retroviruses are not "slow" viruses, as they are frequently termed with regard to their presumed role in carcinogenesis. The "lentiviruses" that are considered models of slow viral pathogenesis (13), but not carcinogenesis, are no exception. Recently an ovine lentivirus known as visna or maedi virus was shown to cause rapid lymphoid interstitial pneumonia in newborn sheep, several weeks after infection (269). This study pointed out that the virus, if expressed at high titer, is directly and rapidly pathogenic. Slow disease may reflect persistent virus expression at restricted sites.
Late Oncogenesis. Since retroviruses without onc genes do not transform cells in culture, all measurements of the latent period of viral oncogenesis are based on studies of infected animals or humans (Table 1). Typically, the latent periods are dated from the time of virus infection and thus are somewhat presumptuous, in that the assumption is made that tumors, if they appear, were initiated by the virus.
The latent period between experimental or congenital infection and lymphomatosis in chickens ranges from 6 months to several years (13, 16, 30, 32, 36, 107). In mice congenitally or experimentally infected with murine leukemia viruses, leukemia takes 6 to 24 months to appear (9, 39, 42, 108). The latent period of mammary carcinomagenesis in mice infected by milk-transmitted MMTV ranges from 6 to 18 months and typically requires several pregnancies of the mouse (16, 54). Longer latent periods of up to 24 months are observed in mice that do not express virus in their milk (55, 109).
The latent period between experimental infection and leukemia is 8 and 12 months in most cats, but only 2 to 3 months in some (62, 66, 90). (The early tumors may have been hyperplasias or tumors induced by feline sarcoma viruses.) The latent period estimated between natural virus infection and leukemia is estimated to be 2 to 3 years in cats that express virus and about 2 to 6 years in cats that do not express virus (63, 66, 110). By contrast, induction of antiviral immunity occurs within several weeks after infection (64, 67).
Bovine leukemia virus-associated leukemias are never seen in animals less than 2 years old and appear at a mean age of 6 years (16). The only experimental bovine lymphosarcoma on record appeared 7 years (73) and some experimental ovine leukemias appeared 4 years (75) after virus inoculation. By contrast, antibody to viral core and envelope proteins appears 4 and 9 weeks after infection (73). Experimental infection of gibbon apes generated leukemia after a latent period of 1 year compared to only 2 weeks for the appearance of antiviral immunity (16, 70).
The latent period for the development of human T-cell leukemia in HTLV-1 positive cancers has been estimated at 5 to 10 years based on the lag between the onset of leukemia and the first appearance of antiviral antibodies of proviral DNA (13, 111, 112). More recently, the latent period of HTLV-I has been raised to record heights of 30 (270) and 40 years (271). By contrast, the latent period of infection and subsequent antiviral immunity was determined to be only 50 days based on seroconversion of the recipients of HTLV-I-positive blood transfusions (81).
The 5- to 40-year latencies claimed for leukemogenesis by HTLV-I are perhaps the most bizarre efforts in linking a virus with a disease. If correct this means either that an infected T-cell becomes leukemic by the time it is 5 to 40 years old or that one of its offspring becomes leukemic in the 50th to 500th generation, assuming an average generation time of a month (176). Clearly the role of the virus in such a process, if any, must be highly indirect. Since all viral genes are essential for replication (13, 204), there is nothing new that the virus could contribute after one round of infection or 24 to 48 hours. This is specifically for HTLV-I and bovine leukemia viruses which are biochemically inactive not only during the long latent period but also during the lethal period of the disease (Sections A and D).
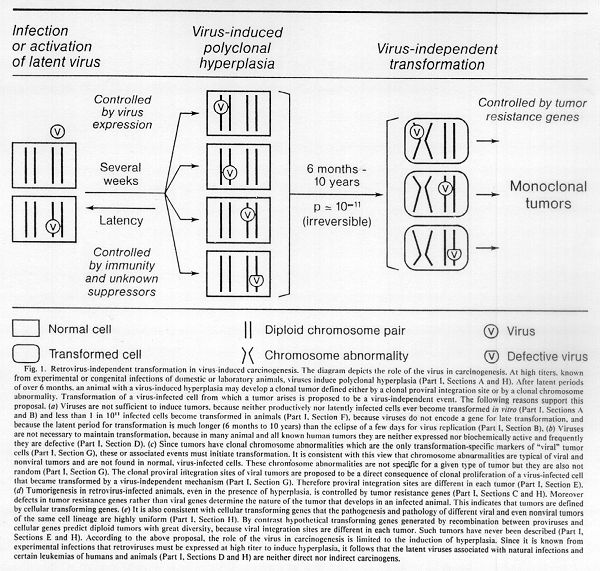
The monumental discrepancies between the long latent periods from 6 months to 10 years for leukemogenesis compared to the short latent periods of several weeks for virus replication or direct pathogenic and immunogenic effects are unambiguous signals that the viruses are not sufficient to initiate leukemia and other tumors (Fig. 1). The viruses are fast and efficient immunogens or pathogens but are either not or are highly indirect carcinogens.
Transformation in Vitro by HTLV-I in 30 to 60 days? Immortalization of primary human lymphocytes infected by HTLV-I or ATLV or simian retroviruses in vitro has been suggested to be equivalent to leukemogenic transformation in vivo (13, 27, 113, 114). If correct, this would be the only example of a retrovirus without onc genes capable of malignant transformation in vitro. The assay infects about 5 x 10 (6th power) primary human lymphocytes with HTLV-I. However, less than one of these cells survives the incubation period of 30 to 60 days, termed "crisis" because the resulting immortal cells are monoclonal with regard to the proviral integration site and because only 4 of 23 such experiments generate immortal cells (115). Since no virus expression is observed during the critical selection period of the immortal cell and since some immortalized cells contain only defective proviruses (115), immortalization is not a viral gene function. Further it is unlikely that the integration site of the provirus (Sections E, G, and H) is relevant to the process of immortalization, since different lines have different integration sites (115). Indeed, spontaneous transformation or immortalization of primary human lymphocytes has been reported applying this assay to simian viruses (113). It follows that immortalization in culture of cells infected by HTLV-I is an extremely rare, perhaps spontaneous event.
There are several indications that in vitro immortalization and leukemic transformation are different events and that both do not depend on HTLV-I: (a) the latent period for immortalization is 30 to 60 days, while that of leukemogenesis is estimated to be 5 to 10 years; (b) in vitro immortalized cells are diploid (116), while all leukemic cells have chromosome abnormalities (Section G); (c ) leukemic cells do not express virus (Section D) while immortalized cells do (115); (d) cells that are clonal with regard to viral integration sites are not necessarily leukemic, because normal T-lymphocytes monoclonal with regard to HTLV-I integration were observed in 13 nonleukemic Japanese carriers (112); (e) finally immortalized cell lines with defective viruses (115) or no viruses (113) indicate that immortalization is a virus-independent, spontaneous event. The evidence that cat, rat, and rabbit cells are immortalized, although they are presumably insusceptible to the human virus (13), endorses this view. It would appear that HTLV-I is directly involved neither in immortalization nor in transformation (Sections A, B, G and H). Instead the assay appears to be a direct measure of cell death of human lymphocytes, due in part to HTLV-I-mediated fusion in vitro (105, 106), and of rare spontaneous immortalization.
C. Tumor Resistance Genes That Inhibit Tumorigenesis but not Virus Replication
If the virus were a direct and specific cause of tumori-genesis, one would expect that all individuals who are permissive for infection would also be permissive for viral tumors. However, this does not appear to be so. For example certain inbred lines of chicken like line 7 (117, 118) or line SC (35, 107) are highly susceptible to induction of lymphoma-tosis by avian retroviruses, whereas line 151 (32, 119, 120) is highly susceptible to induction of erythroblastosis by the same avian retroviruses. By contrast other lines like line 6 (118, 121), line FP (107), or line K28 (122) are either completely or highly resistant to these leukemias but are just as susceptible to virus infection and replication as the tumor-susceptible lines (32, 117, 118, 122, 123). Indeed, both the lymphoma-susceptible SC chickens and the resistant FP chickens develop early viremias and hyperplastic B-cell follicles, but only 50% of the SC chickens develop lymphomas (35, 36). Lymphoma resistance is dominant, indicating that tumor suppressors are encoded (120, 124). The same genes also appear to impart resistance to Rous sarcoma (124). By contrast resistance to erythroblastosis is recessive (Section E).
Analogous tumor resistance genes have been observed in mouse strains. For instance, resistance of C57BL mice to radiation leukemic virus-induced leukemia (125) or of AKR X BALB/c mice to AKR virus-induced leukemia (40) is controlled by the H-2D gene, which is dominant for resistance. Inoculation of the virus into adult C57BL mice caused polyclonal B- and T-cell hyperplasia from which most animals died after 4 to 5 months. However, no leukemia was observed (47). Clearly the tumor resistance genes of the C57BL mice do not suppress virus replication but apparently proliferation of transformed cells. Likewise the SI and the Fv-2 genes of mice inhibit leukemogenesis but not replication of Friend leukemia virus (13, 16, 126). The fates of DBA/2 and ST/b mice inoculated neonatally with AKR virus are another example. After expressing virus for at least 8 months (41), only ST/b mice show a high incidence (about 80%) of leukemia between 8 and 12 months of age, whereas DBA/2 mice show a lower incidence (about 30%) but only at 2 to 3 years of age. Furthermore, not a single lymphomania developed during a period of 1 year in chronically viremic CBA/N mice, inoculated as newborns with Moloney leukemia virus, signalling an absolute resistance to leukemogenesis (42, 46). By contrast, about 90% of viremic AKR mice develop leukemia (40, 48). The wide range of sucsceptibilities to virus-induced leukemia among different mouse strains inoculated with AKR virus, as originally observed by Gross (9), probably also reflects postinfection tumor resistance genes in addition to genes conferring resistance to virus infection and expression (16).
The over 100-fold variation (from less than 1% to 90%) in the incidence of mammary carcinomas among mice that are susceptible to the mammary tumor virus and also contain endogenous MMTVs also reflects host genetic factors that govern resistance to tumori-genesis (16, 54, 55, 58, 127-129). One set of resistance genes governs virus expression, as for example the sex of the host, because almost only females secrete virus and develop tumors (13, 16). Another set governs resistance to carcinogenesis because virus-induced hyperplasia does not necessarily lead to mammary tumors (56, 57).
Resistance to tumorigenesis in animals which are permissive for virus replication indicates that tumors contain nonviral, cellular determinants or tumor antigens. Moreover defects of tumor resistance genes rather than viral genes determine tumor specificity since the nature of the tumor induced by a given virus depends on the host and not on the virus. This lends new support to the conclusion that viruses are not direct causes of tumorigenesis.
D. Tumors without Virus Expression, without Complete Viruses, or without Viruses: Proof that Virus Is Not Necessary to Maintain Transformation
If the retroviruses encode transformation-specific functions, one would expect that viral genes are continuously expressed in viral tumors. However, only 50% of virus-induced avian lymphomas express viral RNA (130). In many clonal lymphomatoses of chickens only incomplete or truncated proviruses are found. These defective proviruses lack the 5' half of the genome and hence are unable to express any viral gene (36, 50, 131, 132).
Moreover neither exogenous nor active endogenous retroviruses can be detected in some lymphomas. One rare study that investigated lymphomatosis in lymphomatosis virus-free chickens found that 10 of about 2000 (0.5%) chickens of line 7 died from lymphomas that were indistinguishable from viral lymphomas at the ages of 6 to 18 months (49, 121). Thus the incidence of lymphoma in virus-free chickens is very similar if not the same as that of chickens infected by lymphomatosis virus with antiviral immunity (less than 1%) (Section A). Since almost all chickens contain multiple endogenous retroviruses (16, 133), it may be argued that these viruses were responsible for the leukemias in animals free of exogenous virus. However, the evidence that endogenous viruses were latent in leukemic as in nonleukemic birds indicated that the endogenous retroviruses were not involved in these spontaneous lymphomas (121). The existence of endogenous viruses in the lymphatoma-resistant chickens of line 6 supports this view (121, 133). In fact, it has been argued that endogenous viruses protect by interference against infection by exogenous variants (13, 16, 134).
A few cases of mouse T-cell lymphomas with defective leukemia viruses have also been observed (135-137). These findings indicate that murine leukemia can also be maintained without expression of retroviral genes.
Expression of mammary tumor virus appears also not necessary to maintain tumors, because no viral antigens (138) and no virus expression are detectable in many virus-positive mammary tumors (9, 52, 139) and because defective proviruses are observed in some tumors (140). Moreover, in mice which lack mammary tumor virus altogether, mammary tumors were observed that cannot be distinguished from virus-positive tumors, indicating that the virus is not necessary to initiate mouse mammary tumors (141). However, in the absence of virus expression, mammary carcinomas develop at lower incidence and after longer latent periods (9, 16, 52, 139-142).
Among virus-positive feline leukemias, some contain only defective proviruses, as in the avian system (143-145). However, about 25 to 35% of all feline leukemias are free of virus, viral antigens (67, 68, 110), and proviral DNA (143-145). This is significantly higher than the percentage of virus-free avian lymphomas. In some virus-free leukemias, the presumably lymphotropic virus is believed to be in other cells of the cat (65).
In provirus-positive natural bovine and experimental ovine leukemias expression neither of virus nor of viral RNA have been detected (75, 146). This result is at odds with the proposal, based on in vitro evidence, that the virus encodes a protein that activates virus transcription and expression of latent cellular transforming genes (147). In addition, the 5' half of bovine leukemia provirus is absent from 25% of bovine leukemias (146, 148). This entirely prevents expression of all viral genes. Other investigators have described that 30% of bovine leukemias are virus free (72).
The proviruses of HTLV-I associated with human T-cell leukemias are also consistently latent. For instance, no expression of viral antigens (149) and no transcription of viral RNA are observed in freshly isolated leukemic T-cells from (5 of 6) HTLV-I positive patients with human T-cell leukemia (150, 151). Again, this is incompatible with the in vitro evidence for a viral transcriptional activator that was proposed to activate virus expression and expression of latent cellular transforming genes (152, 153) (Section H). Moreover, about 10% of the ATLV- or HTLV-I-positive adult T-cell leukemias from Japan contain only defective viruses (77, 151, 154). Since the 5' half of the viral genome was reported to be missing no viral gene expression is possible (77, 151, 155). Further, a minority of Japanese ATL patients appears to be free of ATLV, based on the serological assays that are used to detect the virus (156, 157). A recent analysis found 5 virus-free cases among 69 Japanese ATL patients, who lacked both HTLV-I provirus and antiviral immunity (158). Comparisons among T-cell leukemias in Italy found only 2 of 68 (159) or 3 of 16 (160) otherwise identical cases to be HTLV-I positive. A survey from Hungary found 2 of 326 leukemias antibody positive (161). Other studies from the United States and Italy describe HTLV-I-free T-cell leukemias that share chromosome abnormalities with viral leukemias (Section H). Thus, the ratio of nonviral to viral T-cell leukemias in humans outside Japan appears to be even higher than that of nonviral to viral feline and bovine leukemias.
Since retrovirus expression is not observed in many virus-positive leukemias and since only defective viruses are associated with some leukemias it follows that viral gene products are not necessary to maintain these leukemias. These tumors must be maintained by cellular genes (Section H). The occurrence of "viral" leukemias of chicken, mice, cats, cattle, and humans despite antiviral immunity (Section A) supports this conclusion. This conclusion is also consistent with the evidence that about 30% of the natural feline and bovine leukemias as well as many human and some avian leukemias and murine mammary carcinomas are virus free, yet these tumors cannot be distinguished from viral.
E. Transformation Not Dependent on Specific Proviral Integration Sites
Since retroviruses without onc genes are not sufficient to cause tumors and do not encode transformation-specific functions (Sections A-C) but may nevertheless induce experimental tumors (Section A), several hypothetical mechanisms of viral carcino-genesis have been proposed that each require a specific interaction with the host cell (Section H). One of these postulates is that retroviruses without onc genes activate latent cellular cancer genes, termed proto-onc genes, by site-specific proviral integration (13, 16, 130, 162). The proposal is based on structural analogy with retroviral onc genes, which are hybrids of sequences derived from retroviruses and proto-onc genes (5, 19, 20). It is termed downstream promotion hypothesis (130) because the promoter of the 3' long terminal repeat from the provirus is thought to promote transcription of a proto-onc gene downstream.
It is consistent with this hypothesis that leukemias and other tumors from retrovirus-infected animals and humans are typically all monoclonal with regard to the integration sites of the provirus in the host chromosome. However, if one compares different monoclonal tumors of the same cell lineage, different integration sites are found in each individual tumor. This has been documented for retroviral lymphomas of chickens (37, 131, 132), mice (13, 163, 164), cats (143-145), cattle (146, 148), and humans (13, 151, 154, 155, 165) and also for mammary tumors of mice (13). It is unlikely that the mutant genes generated by provirus integrations are transforming genes, because they are not specific and not known to have transforming function upon transfection. Instead the clonal proviral integration sites of individual tumors appear to be the consequence of clonal proliferation of a single transformed cell from which the clonal tumor originated (Section G).
Relevance of Preferred Integration Regions. Although the search for specific proviral integration sites in viral tumors has met with no success, preferred integration regions were observed in three systems, namely in erythro-blastoses and lymphomas of chicken strains predisposed to these tumors and in mammary tumors of mice bred for susceptibility to this tumor (13, 16). For instance in erythroblastosis-prone 15I chickens that suffer 80% erythroblastosis upon infection (120), integration upstream of proto-erb was observed in 90% (119) and 45% (120, 122) of erythroblastoses. Proto-erb is a proto-onc gene because it is the cellular progenitor of the transforming gene of avian erythroblastosis virus (13, 19). This region-specific integration appears to activate proto-erb transcription compared to certain normal controls (119). However, there are as yet no data on activation of proto-erb translation in leukemic cells. Unexpectedly 45% of the erythroblastoses observed in 15I chickens contained viruses with transduced proto-erb (122). The outstanding yield of proto-erb transductions in this line of chicken compared to others (5, 19) (Section H) suggests an altered proto-erb gene, perhaps already flanked by defective proviral elements which would permit transduction via homologous recombination. It is consistent with this view that in 15I chickens susceptibility in erythroblastosis is dominant (120), while typically resistance to tumors is dominant in chickens and mice (Section C).
Further in about 85% of the viral lymphomas of lymphoma-prone chicken lines (Section C) transcription of the proto-myc gene is activated compared to certain controls (130). Proto-myc is a proto-onc gene because it is the cellular progenitor of the transforming genes of four avian carcinomas viruses, MC29, MH2, CMII, and OK10 (5, 13, 19). Transcriptional myc activation ranges from 300- to 500-fold in some lymphoma lines (RP) to 30-to 100-fold in most primary lymphomas (85%) down to undetectable levels in a few (6%) primary lymphomas (130). However, the activation of proto-myc translation, compared to normal fibroblasts, was estimated as only 7-fold in one RP lymphoma line and even lower in three other lines (166). Assuming that the same ratios of transcriptional to translational activation apply to all lymphomas, activation of myc translation would be only 1- to 2-fold in most lymphomas, hardly enough to explain carcinogenesis. In 5 to 15% of the lymphomas there is no detectable transcriptional activation of proto-myc and the retroviruses appear to be integrated outside of and in random orientation relative to the proto-myc genes (50, 105, 130, 132, 167, 168, 169).
Thus, in lymphomas, proto-myc transcription is frequently but not always activated whereas proto-myc translation appears to be barely, if at all activated. It is not known whether translation of proto-erb is activated in viral erythroblastoses. By contrast viral myc and erb genes are efficiently translated in all virus-transformed cells (5, 13, 16, 19, 20). Moreover in contrast to the hypothetical lymphoma specificity of activated proto-myc, viral myc genes typically cause carcinomas and viral erb genes cause sarcomas in addition to erythroblastosis (5, 13).
Integration of mostly intact murine leukemia viruses into or upstream of proto-myc is also observed in mouse and rat lymphomas. But since it occurs only in 10 (170, 171) to 65% (172) of the cases analyzed, it is not necessary for lymphoma-genesis. Moreover provirus integration near murine proto-myc is also not sufficient for leukemogenesis. Virus integrated near proto-myc was found in 15% of the hyperplastic thymus colonies of AKR mice that appeared 35 days after infection with MCF virus. These colonies were not tumorigenic (172). However, more malignant lymphomas develop from cells with provirus integrated near myc than from other cells, because in 65% of the lymphomas virus was integrated in proto-myc.
There are also preferred regions of provirus integration for MMTV in carcinomas of mice, termed int-1 in C3H mice and int-2 in BR6 mice (13, 16). The int loci or genes are considered to be proto-onc genes only because they are preferred MMTV integration sites. They have not been progenitors of viral onc genes and there is no direct evidence that they can be activated to cellular cancer genes. Moreover transcriptional activation of int is observed only in some tumors (173) and there is no evidence for viral-int hybrid mRNAs (140). It is also not known whether the int loci are coding. The two int loci are totally unrelated to each other and map on different chromosomes (174). Integration within the int regions is neither site nor orientation specific with regard to the int loci (13). Integration at int loci is also not necessary for carcino-genesis, because integration in int-1 is found in only a fraction (22 of 26) of C3H tumors (173) and in int-2 only in a fraction (22 of 45) of BR6 tumors (140). Further integration in int-1 was found in benign hyperplastic nodules that did not become malignant, proving that it is also not sufficient for carcinogenesis (56, 57).
The hypothesis that region-specific integration generates hybrid transforming genes that are equivalent to viral onc genes is inadequate on several counts. (a) Region-specific integration is not necessary for transformation, because in most systems (human, bovine, feline) it is not observed and in all others it is not obligatory. (b) It is also not sufficient for carcinogenesis based on the particular cases of clonal murine leukemia virus integration into proto-myc that did not cause leukemia (172), clonal MMTV integration into int-1 that did not cause mammary carcinomas (56, 57), and monoclonal HTLV-I infections that did not cause T-cell leukemia (112). The non-leukemic proto-myc integration is incompatible with the model purporting that activated proto-myc is like the inevitably transforming viral myc genes (5). The prediction that native proviral-cell DNA hybrids have transforming function, like the related retroviral onc gene models, is unconfirmed. Attempts to demonstrate transforming function of proviral-proto-myc hybrids from chicken lymphomas were negative but led to a DNA with transforming function termed B-lym (13, 175). A plausible reason is that the myc RNAs initiated from upstream viral promoters are poor mRNAs because they start with intron sequences that are not part of normal mRNA and cannot be spliced out, since there is no splice donor downstream of the 3' viral long terminal repeat (Section H). (d) The prediction that the probability of all infected cells to become transformed should be the same as that of region-specific integration is also unconfirmed on the basis of the following calculations (5). The proto-myc, -erb, or int regions that are preferential proviral landing sites in viral tumors measure about 2 and 40 kilobases, respectively (13). Since the chicken chromosome contains about 1 x 10 (6th power) kilobases and the mouse chromosome contains about 3 x 10 (6th power) kilobases, and since provirus integration is random (13, 16), about 2 in 10 (6th power) or 1 in 10 (5th power) infections should generate a tumor cell, if region-specific integration were the mechanism of carcinogenesis. Yet the probability that an infected cell will initiate an monoclonal tumor is only about 10 (-11th power) (Section F). In addition, the latent period of tumorigenesis would be expected to be short because there are at least 10 (8th power) target cells of the respective lineages and many more viruses to infect them (Section F). Moreover, given the long latent periods of carcinogenesis, polyclonal rather than monoclonal tumors would be expected from integrational carcinogenesis. It may be argued that this discrepancy reflects the work of tumor resistance genes. However, postinfection resistance genes that suppress tumor formation by the viral derivatives of proto-myc or erb, like MC29 or avian erythro-blastosis virus, have never been observed in vivo or in vitro. Clearly, since tumor resistance genes do not function in vitro it would be expected that at least 2 of 10 (6th power) cells infected in vitro would be transformed by activation of proto-myc and 2 by activation of proto-erb. However, no transformation by leukemia viruses has ever been observed in vitro (Section B).
In view of this, it is more likely that region-specific integration may provide proliferative advantages to hyperplastic cells or may initiate hyperplasia by activating or inactivating growth control genes rather than being the cause of malignancy. This proposal predicts that integration into proto-myc and proto-erb precedes tumorigenesis (Fig. 1).
It is inconsistent with this proposal that murine leukemia virus integration into proto-myc (172) and MMTV integration into int-1 (56, 57) occur prior to carcinogenesis and thus are not sufficient for carcinogenesis. This proposal predicts also that the chicken lines that are susceptible to lymphoma or erythro-blastosis lack genes that check hyperplasia of lymphocytes or erythroblasts. It is consistent with this view that the same retroviruses cause either lymphomatosis or erythroblastosis or no tumors in different chicken lines. The exclusive (but not absolute) usage of only one of two different int loci by MMTV, namely int-1 in carcinomas of C3H mice and int-2 in BR6 mice, is also more likely to reflect strain-specific activation or inactivation of proliferative controls than two entirely different transforming genes that would nevertheless generate indistinguishable carcinomas.
F. The Probability That a Virus-infected Cell Will Become Transformed Is Only 10 (-11th power)
To calculate the probability that a virus-infected cell will become transformed, we must consider the ratio of symptomatic to asymptomatic carriers, the clonality of the viral tumors, and the long latent periods of oncogenesis. (a) The ratio of symptomatic to asymptomatic carriers with latent infections and antiviral immunity averages less than 10 (-3rd power) (Section A), but that of viremic animals susceptible to transformation may reach 0.9 (Section C). (b) Since monoclonal tumors emerge from at least 10 (8th power) B- or T-cells (176), the probability of an infected cell in an animal to become the progenitor of a clonal leukemia is only about 10 (-8th power). This calculation assumes that all of these cells are infected. This is certainly true for the mice that carry AKR virus, radiation leukemia virus (82), or inducible mammary tumor virus (75, 142) in their germ line, and is probably the case in congenitally infected viremic chickens, cats, gibbons, and mice (12, 16, 31, 39, 63, 66, 70). In fact in viremic animals, the hyperplastic effect of the virus would have enhanced the number of prospective tumor cells to at least 10 (9th power) (Sections A and B). Even if only a fraction of susceptible cells are infected in animals or humans with latent infections and antiviral immunity, the number of infected cells per host is estimated to be at least 10 (6th power) to account for the immune response (Section B, and Refs. 13, 16, 27, 31, and 63) or the proviruses that are used to diagnose latent virus infection (Section D). Proviruses cannot be detected biochemically unless they are present in at least 1 of 100 cells. © Finally, the probability of an infected cell to become transformed in an animal is a function of the number of generations of infected cells that occur during the latent period of the disease. Given latent periods of 6 to 120 months (Section B) and assuming an average life span of 1 month for a susceptible B- or T-cell (176), about 10 to 100 generations of infected cells are required to generate the one transformed cell from which a clonal tumor emerges. The corresponding probability that a generation of cells will develop a clonal tumor would be 10 (-1 power) to 10 (-2 power). Considering the proliferative effect of the virus on hemopoietic target cells in viremic animals, this may again be a conservative estimate. Indeed, a mitotic rate of 1 day has been assumed for B-cells of lymphoma-tosis virus-infected chickens (177).
Thus the probability that a virus-infected, hemopoietic cell will become transformed in an individual with a latent infection and antiviral immunity is about 10 (-3 power) x 10 (-6th power) x 10 (-2 power) = 10 (-11th power), and that in a viremic individual without tumor resistance genes is about the same, namely 0.9 x 10 (-9th power) x 10 (-2nd power) = 10 (-11th power). Therefore the increased risk of viremic animals to develop leukemia must be a direct consequence of the hyperplasia of prospective tumor cells (Section A) (Fig. 1). In tumor-resistant animals the probability that the infected cell will become transformed may be the same, but the resistance genes would prevent proliferation of the transformed cells (Section C and H). The apparent probability that virus-infected, non-hemopoietic cells will become transformed must be lower in both susceptible and resistant animals, because the incidence of solid tumors is much lower than that of leukemia (9, 32).
G. Clonal Chromosome Abnormalities Are the Only Transformation-Specific Markers of Retrovirus-infected Tumor Cells: Causes of Transformation?
The evidence that viral tumors are monoclonal (Section E) and that leukemogenesis by retroviruses (without onc genes) is highly dependent on tumor resistance genes, which are different from genes that determine susceptibility to the virus, suggest virus-independent steps in carcinogenesis (Section C). Indeed clonal chromosome abnormalities of virus-positive mammalian tumors provide direct evidence for cellular events that may be necessary for carcinogenesis. (Avian cells have not been studied because of their complex chromosome structure.) For example, trisomies of chromosomes 15 have been observed frequently in viral T-cell leukemias of mice (16). In addition translocations between chromosomes 15, 17, and others have been recorded (108, 178-180, 272). In mammary carcinomas of mice, a chromosome 13 trisonomy was observed in 15 of 15 cases including inbred GR and C3H mice (which contain MMTV) and outbred Swiss mice (which probably also contain the virus) (181). Clonal chromosome abnormalities have also been observed in 30 of 34 bovine leukemias induced by bovine leukemia virus (75). A recent cytogenetic analysis of human adult T-cell leukemias (ATL) from Japan showed that 10 of 11 cases had an inversion or translocation of chromosome 14 (183). Rearrangements of other chromosomes have been detected in 6 of 6 (184), 12 of 13 (116), and 8 of 9 cases of HTLV-I-positive leukemias (185). Thus over 90% of virus-positive T-cell leukemias have chromosome abnormalities. A survey of all viral T-cell leukemias analyzed shows rearrangements of chromosome 14 in 26% and of chromosome 6 in 29% (186, 187).
The chromosome abnormalities of these viral leukemias and carcinomas are as yet the only known determinants that set apart transformed from normal virus-infected cells. Since the chromosome abnormalities are clonal, the origin of the tumor must have coincided with the origin of the chromosome abnormality. Therefore chromosome abnormalities or closely associated events must be directly relevant to initiation of tumorigenesis. They could either be, or coincide with, a single step mechanism of transformation or with one of several steps in transformation, as postulated in the case of the Philadelphia chromosome (188). It is consistent with this view that chromosome abnormalities are found in all virus-infected tumors analyzed.
However, heterogeneity among the karyotypes of individual human or murine leukemias of the same lineage (16, 179, 182, 189, 190, 272) and thus heterogeneity of mutation support the view that chromosome abnormalities are coincidental with rather than causal for transformation. Yet this view does not take into consideration that together with the microscopic alterations, other submicroscopic mutations may have occurred that could have initiated the disease (108). It is consistent with this view that tumor cells contain in addition to microscopic karyotype changes submicroscopic deletions, detectable as restriction enzyme site polymorphisms (191). Some of these mutations may be functionally equivalent to the truncation-recombination mechanism that activates the docile proto-onc genes of normal cells to the onc genes of directly oncogenic retroviruses (5, 192). Thus specific karyotypic changes may only be the tip of the iceberg of multiple chromosomal mutations, referred to as "genequake," [G. Matioli, personal communication] which must have occurred in the same cell. One or several of these could have initiated the tumor. Chromosome recombination sites are also postulated to be cellular transforming genes of virus-negative tumors, as for example in Burkitt's lymnphoma (5) or in human leukemia with the Philadelphia chromosome (193).
If chromosomal abnormalities are necessary for transformation of cells infected by retroviruses without onc genes, chromosomal abnormalities would not be expected in tumors caused by retroviruses with directly transforming onc genes. This has indeed been confirmed for tumors caused in mice by Rous sarcoma virus (194) or by Abelson leukemia virus (195) which have normal karyotypes (Table 1).
The clonality of retrovirus-positive tumors is then defined in two different ways: by a retroviral integration site (see Section E), and by a chromosome abnormality (see Fig. 1). Each of these two clonal chromosome alterations could then mark the origin of the tumor, while the other must have pre-existed. Since the tumors originate late after infection and probably from a virus-infected, normal cell, the clonal retroviral integration site would appear to be a direct consequence of clonal proliferation of a cell transformed by a chromosome alteration. Indeed chromosome abnormalities are typical of tumor cells but not of virus-infected normal cells. This view is consistent with the evidence that retrovirus integration does not cause transformation and that transformation is not dependent on specific integration sites. It is also highly improbable that chromosome abnormalities are caused by the virus, because they are not found in virus-infected normal cells and because they are also characteristic of virus-negative tumors (Section H). The clonal retroviral integration sites in viral tumors the chromosomes of which have not been analyzed, as for example avian, feline, and simian leukemias, may indeed signal as yet undetected clonal chromosome abnormalities.
Virus-independent Transformation in Virus-positive and -negative Tumors
Several hypotheses postulate that retroviruses play a direct role in carcinogenesis. One reason is that viruses, seemingly consistent with Koch's first postulate, are associated with tumors although frequently in a latent or defective form. In addition it appears consistent with Koch's third postulate that experimental infections with retroviruses may induce leukemia under certain conditions (see Sections B and C). However, none of these hypotheses provide an adequate explanation for the fact that retroviruses are not sufficient to initiate (Sections A to C) and not necessary to maintain (Sections D and E) transformation and do not encode a transformation-specific function. Moreover none of these hypotheses can explain why transformation is initiated with a clonal chromosome abnormality (Section G) and why tumor specificity is determined by the host rather than the virus (Sections C and E). The short-comings of three of these hypotheses are briefly reviewed here.
1. The Oncogene Hypothesis. Huebner (8) and others (9, 82) have postulated that retroviruses (without onc genes) are direct carcinogens that include oncogenes, hence the term "oncogene hypothesis" (8). The hypothesis was based on abundant positive correlations between retrovirus expression and cancer incidence in laboratory mice and domestic chickens, which indeed suggested direct viral etiology in apparent accord with Koch's third postulate. The hypothesis generalized that either import of retroviruses from without, or activation of latent viruses from within, is the direct cause of spontaneous, chemically induced, or physically induced tumors (8, 9, 82). However, the hypothesis failed to account for the long latent periods of oncogenesis and for complete tumor resistance by certain animals that are highly susceptible to the virus and for host genes that would determine tumor specificity (Section C). Above all the hypothesis failed to account for the monoclonality and the chromosome abnormalities of the resulting tumors.
2. The Hypothesis That Latent Cellular Cancer Genes Are Activated by Provirus Integration. This hypothesis has been introduced in Section E. It holds that retroviruses act as direct, albeit inefficient carcinogens by generating hybrid transforming genes from proviruses joint with cellular proto-onc genes. Excepting the specific cases described in Section E, this mechanism makes four clear predictions, namely: (a) that different transforming genes exist in each tumor, because each has a different proviral integration site (Section E); (b) that therefore a large number of tumor resistance genes exist in tumor-resistant animals (Section C); (c ) that provirus-cell hybrid genes are expressed to maintain transformation; and (d) that virus-transformed cells exist without chromosome abnormalities, analogous to cells transformed by retroviruses with onc genes (Section G).
None of these predictions is confirmed, (a) Contrary to the expectation for many different transforming genes, all virus-positive tumors of a given lineage are phenotypically highly uniform (Section A). Even virus-free tumors are indistinguishable from virus-positive tumors of the same lineage only by the presence of viruses. Examples are the identical pathologies and pathogeneses of viral and nonviral murine leukemias (196-198), chicken B-cell lymphomas (121), human T-cell leukemias (158, 161, 186), and mouse mammary tumors (11, 139, 141, 142) (Section D). (b) Contrary to expectation only a small set of cellular resistance genes controls the development of viral tumors in chicken or mice (13, 16) (Section C). Moreover apparently the same resistance genes of chickens of line 6 suppress viral and nonviral lymphomas, and even lymphomas induced by Marek's virus (124). By contrast chickens of line 7 that lack these genes are equally susceptible to both (121) (Section D). Mice provide parallel examples such as in the CBA strain, which is resistant to spontaneous (9) as well as to viral (46) leukemia (Section C). (c ) Contrary to expectation for virus-cell hybrid transforming genes, proviruses are latent or defective and biochemically inactive in many animal and all bovine and human leukemias (Section D). (d) Contrary to expectation for viral carcinogenesis all virus-positive murine, bovine, and human tumors analyzed have chromosome abnormalities. Further, similar chromosome abnormalities in viral and nonviral tumors again suggest common cellular transforming genes. For instance, the same chromosome 15 trisomy is observed in murine leukemias induced by viruses, chemicals, or radiation (180, 190, 199-201, 272). In addition virus-positive and virus-free human T-cell leukemias have common abnormalities in chromosomes 14 and 16 (160, 183, 186, 187, 189, 202, 203). Since all human T-cell leukemias and all bovine leukemias have chromosome abnormalities but not all are infected by viruses (Sections D and G), it would appear more likely that the viruses are coincidental passengers rather than causes of the disease.
3. The Hypothesis That Latent Cellular Cancer Genes Are trans-Activated by Viral Proteins. This hypothesis postulates that certain retroviruses directly activate latent cellular transforming genes with a specific viral protein. This has been proposed for bovine leukemia virus and human HTLV-I based on in vitro models (147, 152, 153) (see Section D). However, the hypothesis is unlikely for the following reasons. Since the putative trans-activation protein of HTLV-I is essential for replication (204), all cells in which the virus replicates would expect to be transformed. This is clearly not the case. Further this gene cannot be relevant for transformation since bovine and human leukemias in particular do not express viral RNA or protein or cannot express RNA or protein because of defective proviruses (Section D). In addition this hypothesis also fails to account for the chromosome abnormalities found in all bovine and human leukemias (Section G). Finally both the proviral insertion and the transactivation hypotheses fail to explain the inevitably long latent periods of viral tumori-genesis (Section B).
Therefore it is proposed that transformation is a virus-independent event that must be due to cellular genes (Fig. 1). These genes would be generated by chromosomal mutations for which chromosome abnormalities are a macroscopic indicator. This explains the clonal chromosome abnormalities that could not be predicted by any of the virus-cancer hypotheses. In a given lineage of cells the number of cellular genes convertible to transforming genes must be limited since they cause highly uniform tumors which can be suppressed by a small set of resistance genes.
Retrovirus-independent transformation resolves the apparent paradox that tumors occur very seldom in typical natural infections of wild animals and humans, and then only long after infection, and despite viral latency and antiviral immunity. It is also consistent with virus-independent transformation that the probability that an individual virus-infected cell will become transformed is only 10 (-11th power) and that this probability is the same in a viremic chicken with a virus-induced hyperplasia, as in a normal chicken with a latent infection and antiviral immunity (Section F). The low probability of virus-independent transformation also explains directly why cells infected by retroviruses are not transformed in culture, namely because not enough cells can be maintained for a long enough time to observe spontaneous transformation. Virus-independent transformation is also compatible with tumor resistance genes that do not inhibit viral replication or growth of normal virus-infected cells. In addition it is consistent with the notion that defects of cellular resistance genes rather than viral genes determine tumor specificity (Section C).
The role of the virus in tumorigenesis is then limited to the induction of hyperplasia by activating cellular proliferative functions either from within or from without via viral antigens or virus-induced growth factors (13, 16, 46). For this purpose the virus must be expressed at a high titer or it must have infected a large number of cells, if insertional mutagenesis of proliferative genes were involved (Section E). This may be similar to the mechanism whereby DNA viruses induce transformation, as for example Epstein-Barr virus which is thought to induce Burkitt's lymphoma. Exactly like their retroviral counterparts, all Burkitt's lymphomas have chromosome abnormalities but not all contain the virus (5). Thus the role of the retrovirus in carcinogenesis is as indirect as that of chemical or physical carcinogens.
Alternatively a latent retrovirus may itself be subject to activation by physical, chemical, or spontaneous events that can induce hyperplasis and cancer (8, 12, 82) (Fig. 1). The physically activated radiation leukemia virus (82) or the chemically activated endogenous retroviruses of mice or chickens (12, 16) are examples. It is uncertain whether under these conditions the retrovirus is just an indicator or an intermediate of proliferative activations that may lead to carcinogenesis because comparable studies with virus-free strains of animals are not available. The physically or chemically inducible phages or herpes viruses may in turn be models for this (11, 83).
Little is known about the nature of the hyperplastic cell. The existence of viral hyperplasias in tumor-resistant animals indicates that the hyperplastic cell is not neoplastic (Section C). Most hyperplastic cells are polyclonal with regard to proviral integration sites (118) and are likely to have a normal karyotype, as has been shown in some cases (47) (Section C). Hyperplastic cells with normal karyotypes have also been observed as precursors of radiation leukemia in mice (205). Nevertheless the evidence for clonality with regard to a proviral integration site in T-cell hyperplasias (172) and mammary hyperplasias (56, 57) of mice and in T-cells of healthy humans (112) indicates clonal, possibly virus-induced alterations that are not sufficient for carcinogenesis. One could speculate then that hyperplastic cells fall into two classes, those which respond to viral antigens delivered from within or without (42) and those which respond to growth control genes altered by provirus integration (Section E).
Notable exceptions to virus-independent transformations are infections that generate retroviral transforming genes. However, the probability of generating a retrovirus with an onc gene is clearly much lower than integration into a cellular gene (10 (-6th power), Section E) and even significantly lower than virus-independent transformation (10 (-11th power), Section F) (273). Only about 50 such viral isolates have been recorded in history (5, 13, 19). (The frequent erb transductions from the chicken 15I line are an exception to this rule (Section E).) The generation of these viruses requires two rare illegitimate recombinations to transduce a transformation-specific sequence from a cell into a retrovirus vector (5, 19, 20, 273). However, one illegitimate recombination that unites the 5' promoter, translational start sequence, and splice donor of a retrovirus with a transformation-specific sequence from a cellular proto-onc gene would be enough to generate a functional virus-cell recombinant onc gene that cannot be replicated. Tumors caused by such genes are presently unknown. They will be harder to diagnose but are probably more frequent than the rare, natural tumors containing complete retroviruses with onc genes (273).
This raises the question of why orthodox integration of a provirus within a proto-onc gene, like proto-myc, is not observed to transform infected cells in vivo or in vitro with the predicted probability. Based on the calculations described in Section E, this probability should be about 1 in 10 (4th power) considering that about 20 proto-onc genes are known from 20 viral onc genes (5, 13, 19). A possible answer is that proviruses abutting proto-onc genes from the proviral ends rather than from within, as in viral onc genes (273), provide neither new downstream translational starts nor splice donors for those coding regions of the proto-onc genes that are separated from their native start signals by the inserted provirus. Nevertheless they can provide efficient downstream promoters (130) of RNAs that may not be translatable.
I. Are Retroviruses a Basis for Cancer Prediction, Prevention, or Therapy?
In assessing the tumor risk of a retrovirus-infected animal or human, latent infections must be clearly seperated from chronic, acute, or viremic infections. The control of virus expression in a given host is a product of three factors: the virus; the host cell; and the animal. The viral factor is defined by viral genes and promotors (13, 16, 206). The cellular factor is defined by genes that encode viral receptors and unknown suppressors (8, 9, 11-13, 16-18, 82). The animal factor is defined by antiviral immunity.
By far the most common natural retrovirus infections are latent, chronic infections that persist in animals and humans in the presence of antiviral immunity presumably only in a limited number of cells (38, 40, 90, 207). The leucemia risk of this statistically most relevant group of natural infections avarages about less than 0.1% in different animal species (Section A). It is possibly the same as, but certainly not much higher than, that of uninfected controls (Sections A and D). Thus latent viruses offer no targets for tumor prevention. The low probability that an immunocompetent individual will develop chronic viremia and hence leukemia also suggests that retroviruses carrying therapeutic genes are not a significant risk as leukemogens.
By contrast the leukemia risk of a viremic animal that survives the early pathogenic effects of the infection (Section B) can be high barring tumor-resistance genes (Section A and C). It ranges between 0 and 90% in different lines of chicken or strains of inbred mice and avarages about 30% in domestic cats. However, outside the laboratory chronic viremias are very rare and have never been recorded in humans. They result either from congenital infections in the absence of maternal antibody (Section A) or from rare, native immunodeficiency (66).
Thus a predictable tumor risk depends entirely on high virus expression and virus-induced hyperplasia. This risk can be reduced or prevented by limiting or blocking lymphoblast hyperplasia as for example by bursectomy or thymectomy (Section A). Alternatively, inoculation of newborn AKR mice with antiviral antibody was observed to suppress viremia and subsequent leukemia in 68% (208). It would appear more practical, however, to breed or select animals with genes that confer resistance either to the virus or tumorigenesis or both.
Above all, neither active nor latent viruses offer targets for tumor therapy, since tumors are not maintained and are not directly initiated by viral genes, and also occur despite active antiviral immunity.
Clearly the cell is the more complex variable in the as yet poorly defined interaction between retroviruses and cells that leads to hyperplasia and than carcinogenesis. In view of the evidence for cellular genes that determine resistance to hyperplasia and tumorgenesis, further progress in understanding and treating virus-induced cancer will depend on identifying cellular determinants of carcinogenesis and the function of hyperplasia and tumor resistance genes.
II. Retroviruses and AIDS
The isolation in 1983 of a retrovirus from a human patient with lymphoadenopathy, a typical symptom of AIDS, led to the proposal that the virus, now termed lymphadenopathy-associated virus, is the cause of AIDS (26). Related viruses, termed HTLV-III, ARV, or HIV (209), have since been isolated from about one-half of the AIDS patients that have been sampled (210-214). In the United States about 26,000 AIDS cases and 15,000 AIDS fatalities have been reported between 1981, when the disease was first identified (215), and October 1986 (216). Women represent only 7% of the AIDS cases in the United States (216). The number of AIDS cases reported in the United States has increased from about 100 per 6-month period in 1981 to about 5,000 during the last three 6-month periods from January 1985 (216). At the same time the case fatality rate has declined from a high of 88% in 1981 to 32% in 1986 (216). In absolute numbers the known deaths have declined from a high of 2,600 in the first 6 months of 1985 to 1,800 in the first 6 months of 1986. This suggests either that the virulence of the disease is dropping or that other diseases were diagnosed as AIDS. Recently the virus was also suggested to cause disease of the brain and of the nervous system (230, 255, 268, 274) and lymphoid interstitial pneumonia (275).
Antibody to the virus is found in about 90% of AIDS patients and correlates with chronic latent infection by the virus (217-221). Because of the nearly complete correlation between AIDS and immunity against the virus, the virus is generally assumed to be the cause of AIDS (13, 27). Accordingly, detection of antiviral antibody, rather than virus, is now most frequently used to diagnose AIDS and those at risk for AIDS (27, 217-224). This is paradoxical, since serum antibody from AIDS patients neutralizes AIDS virus (225-227) and since antiviral immunity or vaccination typically protects against viral disease. It is even more paradoxical that a low antibody titer is equated with a low risk for AIDS (228, 229).
Unlike all other retroviruses, AIDS viruses are thought to be direct pathogens that kill their host cells, namely T-lymphocytes (13, 27), and possibly cells of the brain (230, 255). This view is compatible with the phenotype of AIDS, the hallmark of which is a defect in T-cells (13, 27, 215), and with experimental evidence that many but not all viral isolates induce cytopathic fusion of T-lymphocytes under certain conditions in vitro (Section D). Further it is incompatible with neurological disease (231, 232, 255). However, cell killing is incompatible with the obligatory requirement of mitosis for retrovirus replication (16, 25) and with the complete absence of cytocidal effects in all asymptomatic infections in vivo (Section D).
A. Infections with No Risk and Low Risk for AIDS Indicate That the Virus Is Not Sufficient to Cause AIDS
Since their original discoveries in AIDS patients, the virus and more frequently antibody to the virus have also been demonstrated in a large group of asymptomatic persons (212, 214). The virus has been estimated to occur in about 1 to 2 x 10 (6th power) or about 0.5 to 1% of all Americans (223, 224). In the United States persons at high risk for infection include promiscuous homosexual and bisexual men, of whom 17 to 67% are antibody positive; intravenous drug users, of whom 50 to 87% are positive; and hemophiliacs, of whom 72 to 85% are positive according to some studies (13, 218, 223). On the basis of this particular epidemiology, it was concluded that the virus is not transmitted as cell-free agent like pathogenic viruses but only by contacts that involve exchange of cells (13, 27).
In these virus-infected groups the annual incidence of AIDS was found to average 0.3% (224) and to reach peak values of 2 to 5% (218, 223, 233). However even in these groups there are many more asymptomatic than symptomatic virus carriers.
Other infected groups appear to be at no risk for AIDS. In Haiti and in certain countries in Africa antibody-positive individuals range from 4 to 20% of the population, whereas the incidence of AIDS is estimated at less than 0.01% (223, 229, 234). Several reports describe large samples of children from Africa who were 20 (228) to 60% (221) antibody positive and of female prostitutes who were 66 to 80% antibody positive (221, 235), yet none of these had AIDS. Among male homosexuals and hemophiliacs of Hungary about 5% are AIDS virus positive, yet no symptoms of AIDS were recorded (161). Among native male and female Indians of Venezuela 3.3 to 13.3% have antiviral immunity, but none have symptoms of AIDS (236). Since these Indians are totally isolated from the rest of the country, in which only one hemophiliac was reported to be virus positive (236), the asymptomatic nature of their infections is not likely to be a consequence of a recent introduction of the virus into their population. Thus it is not probable that these infections will produce AIDS after the average latent period of 5 years (Section B).
Since the percentage of virus carriers with symptoms of AIDS is low and in particular since it varies between 0 and 5% depending on the AIDS risk group of the carrier, it is concluded that the virus is not sufficient to cause AIDS and that it does not encode an AIDS-specific function. The virus is also not sufficient to cause neurological disease, since it has been detected in the brains of persons without neurological disease and of healthy persons who had survived transient meningitis (230-232).
Thus the virus appears only rarely compatible with Koch's third postulate as an etiological agent of AIDS. It may be argued that the asymptomatic infections reflect latent infections or infections of only a small percentage of susceptible cells, compared to presumably acute infections with symptoms of AIDS. However, it is shown in Section C that infections of neither symptomatic nor asymptomatic carriers are acute; instead both are equally latent and limited to a small percentage of susceptible cells.
Further the observations that some virus carriers are at high and others at essentially no risk for AIDS directly argue for a cofactor (218, 237) or else for a different cause for AIDS. The strong bias against women, because only 2.5% (479 of 17,000 cases) of the sexually transmitted AIDS cases in the United States are women (216), is a case in point. The virus-positive but AIDS-negative children and prostitutes of Africa (221) or Indians from Venezuela (236) are other examples.
B. Long Latent Period of AIDS Incompatible with Short Latent Period of Virus Replication
The eclipse period of AIDS virus replication in cell culture is on the order of several days, very much like that of other retroviruses (238). In humans virus infection of a sufficient number of cells to elicit an antibody response appears to take less than 4 to 7 weeks. This estimate is based on an accidental needle-stick infection of a nurse, who developed antibody 7 weeks later (239), and on reports describing 12 (240) and 1 (232) cases of male homosexuals who developed antibody 1 to 8 weeks after infection. During this period a mononucleosis-like illness associated with transient lymphoadenopathy was observed. In contrast to AIDS (see below), this illness appeared 1 to 8 weeks after infection and lasted only 1 to 2 weeks until antiviral immunity was established. The same early mononucle-osis-like disease, associated with lymphocyte hyperplasia, was observed by others in primary AIDS virus infections (234). This is reminiscent of the direct, early pathogenic effects observed in animals infected with retroviruses prior to the onset of antiviral immunity (Part I, Section B).
By contrast the lag between infection and the appearance of AIDS is estimated from transfusion-associated AIDS to be 2 to 7 years in adults (220, 223, 241, 242) and 1 to 2 years in children from infected mothers (220, 223). The most likely mean latent period was estimated to be 5 years in adults (220, 223). Unexpectedly, most of the AIDS virus-positive blood donors identified in transfusion-associated AIDS transmission did not have AIDS when they donated blood and were reported to be in good health 6 years after the donation (220). Likewise there is evidence that individuals shown to be antibody positive since 1972 have not developed AIDS (228). Further 16 mothers of babies with AIDS did not have AIDS at the time of delivery but three of them developed AIDS years later (276). This indicates that the latent period may be longer than 5 years or that AIDS is not an obligatory consequence of infection.
In view of the claim that the virus directly kills T-cells and requires 5 years to cause disease, we are faced with two bizarre options: Either 5 year old T-cells die 5 years after infection or the offspring of originally infected T-cells die in their 50th generation, assuming a generation time of one month for an average T-cell (176). It may be argued that the virus is biochemically inactive during the first five years of infection and then activated by an unknown cause. However, AIDS virus is biochemically inactive even during the acute phase of the disease (Section C). Moreover it would be difficult for the retrovirus to become acute five years after it had induced chronic antiviral immunity.
Because of the 5 year latency between infection and AIDS, the virus has been likened to the lentiviruses (277), a group of animal retroviruses that is thought to cause debilitating diseases only after long latent periods (13) (Part I, Section B). However recently an ovine lentivirus, the visna or maedi virus of sheep, was shown to cause lymphoid interstitial pneumonia in 2 to 4 weeks if expressed at a high titer (269). (The same disease is believed to be caused by AIDS virus in humans (see below)). Therefore lentiviruses are not models for retroviruses that are only pathogenic after long latency (Part I, Section B).
Based on the 5-year latent period of the disease and on the assumption that virus infection is sufficient to cause AIDS, one would expect the number of AIDS cases to increase to 1 to 2 x 10 (6th power) in the United States in the next 5 years. The virus has reportedly reached its present endemic level of 1 to 2 x 10 (6th power) in the United States (223, 224) since it was introduced there, presumably, less than 10 years ago (27). Yet the spread of AIDS from 1981 to 1986 has not followed the spread of virus with a latent period of 5 years. Instead, recent statistics (see above) indicate no further increases in the number of AIDS cases and a significant decline in the number of AIDS fatalities in the United States (216, 244).
Clearly, the long lag between infection and AIDS and the large number of virus-positive cases in which as yet no AIDS is observed, even after long latent periods, lead to the conclusion that the virus is not sufficient to induce AIDS and does not encode an AIDS-specific function. Indeed, this conclusion is directly supported by genetic evidence against a viral AIDS gene. Deletion analysis has proved that all viral genes are essential for replication (28, 245), which requires not more than 1 or 2 days, yet AIDS follows infection only with an average lag of 5 years and even then only very rarely.
C. Levels of AIDS Virus Expression and Infiltration Appear Too Low to Account for AIDS or Other Diseases
If AIDS viruses were pathogenic by killing susceptible lymphocytes, one would expect AIDS to correlate with high levels of virus infiltration and expression, because uninfected cells would not be killed by viruses nor would unexpressed or latent viruses kill cells. As yet no report on virus titers of AIDS patients has appeared, despite the record interest in the epidemiology and nucleic acid structure of this virus (13, 27, 223). In view of the consistent antiviral immunity of AIDS patients and the difficulties in isolating virus from them (213), the virus titers are probably low. Titers have been said to range between only 0 and 10 (2nd power) per ml blood (213, J.A. Levy, personal communication.)
Proviral DNA has been detected in only 15% (9 of 65) AIDS patients; in the remaining 85% the concentration of provirus, if present, was apparently too low for biochemical detection (246). Viral RNA was detected in 50 to 80% of AIDS blood samples. However, among the positive samples, RNA was found in only less than 1 of 10 (4th power) to 10 (5th power) presumably susceptible lymphocytes (247). The relatively high ratios of provirus-positive (10 (-2 power) to 10 (-3 power)) to viral RNA-positive cells (10 (-4 power) to 10 (-5 power)) of AIDS patients indicate latent infections. Further there is no evidence that the virus titer or the level of virus infiltration increases during the acute phase of the disease. It is probably for this reason that cells from AIDS patients must be propagated several weeks in culture, apart from the host's immune system, before either spontaneous (210-214) or chemically induced (248) virus expression may occur. Further, the AIDS virus is completely absent from the Kaposi sarcoma (27, 246), which is associated with 15% (216) to 30% (249) of AIDS cases and is one of the most characteristic symptoms of the disease.
Similar extremely low levels of virus infiltration and expression were also recorded in AIDS virus-associated brain disease (274). Likewise, in interstitial lymphoid pneumonia less than 0.1% of lung cells expressed viral RNA (275). Indeed there is evidence that even latent virus may not be necessary for AIDS, since 85% of AIDS patients lack proviral DNA (246) and since over 10% of AIDS patients have been observed to lack antiviral immunity (214, 221, 222, 234). Further, in a study from Germany 3 of 91 AIDS patients were found to be virus free, based on repeated negative efforts to detect antibody or to rescue virus. [H. Ruebsamen-Waigmann, personal communication.]
It is concluded then that the AIDS virus infects less than 1%, and is expressed in less than 0.01%, of susceptible cells both in carriers with or without AIDS. This raises the question of how the virus could possibly be pathogenic and responsible for immunodeficiency or other diseases. For instance even if the virus were to claim its 10 (-4th power) or 10 (-5th power) share of T-cells that express viral RNA every 24 to 48h, the known eclipse period of retroviruses, it would hardly ever match or beat the natural rate of T-cell regeneration (176).
All other viruses function as direct pathogens only if they are biochemically active and expressed at high levels. For instance, the titers that correlate with direct pathogenicity for avian retroviruses are 10 (5th to 12th power) (31, 35, 250) and they are 10 (4th to 7th power) for murine retroviruses (12, 38, 40, 42, 251) (Section B). Hepatitis viruses reach titers of 10 (12th to 13th power) when they cause hepatitis (15), and latent infections are not pathogenic (83). Further, the very low levels of AIDS virus expression in vivo are difficult to reconcile with reports based on in vitro studies with synthetic indicator genes that the AIDS virus encodes a potent transcription-stimulating protein (28, 153, 245). Clearly such activators are not at work in vivo.
The extremely low virus titers of symptomatic and asymptomatic carriers also explain why infection by the virus in the United States is essentially limited to contacts that involve transmission of cells (244) rather than being transmitted as a cell-free, infectious agent like pathogenic viruses. For instance, among 1750 health care workers with exposure to AIDS, only 1 or 2 were found to be antibody positive (252). Another study failed to find a single antibody-positive person among 101 family contacts of 39 AIDS patients, all of whom had lived in the same household with an AIDS patient for at least 3 months (253).
D. AIDS Viruses Not Directly Cytocidal
The AIDS viruses are reported to display in culture a fast cytocidal effect on primary T-cells within 1 to 2 months after infection (13, 27, 254). The cytocidal effect was shown to involve cell fusion (27, 238, 254). The effect is thought to reflect the mechanism of how the virus generates AIDS after a latent period of 5 years (27, 254).
This is debatable on several grounds: (a) above all, the in vitro assay cannot account for the large discrepancy between the short latent period of cell death in vitro and the 5-year latent period of the disease; (b) T-cell fusion is not observed in vivo in chronic, asymptomatic virus carriers and not in prospective AIDS patients during the long latent period of the disease (255), although virus expression is not lower than during the acute phase of AIDS; © T-cell killing is also not observed in T-cell lines in vitro (27) and not in primary lymphocytes under appropriate conditions (238). Further primary lymphocytes infected by AIDS virus were shown to double every 5 days in cell culture for three weeks; at the same time the previously latent AIDS virus was activated to high levels of expression (278); (d) virus strains that do not cause cytopathic fusion in vitro have been isolated from 7 of 150 AIDS patients. [H. Ruebsamen-Waigmann, personal communication.] This demonstrates that the fusion-inducing function of the virus can be dissociated from a putative AIDS function.
Thus T-cell killing by fusion is apparently a cell culture artifact that depends on the virus strain and the cell used, as has been shown for many other retroviruses including HTLV-I (Part I, Section B), and not an obligatory feature of virus infection. As with other retroviruses, fusion involves binding of viral envelope antigens on the surface of infected cells with receptors of uninfected cells. Accordingly, fusion is inhibited by AIDS virus-neutralizing antibody (256). It apparently depends on high local virus titers that in particular in the case of AIDS are not observed in vivo. This view of the cell-killing effect also resolves the apparent contradiction between the postulated cytocidal effects of AIDS viruses and the obligatory requirement of all retroviruses for mitosis in order to replicate (16, 25). Indeed AIDS viruses have been reported to replicate without cytocidal effects not only in T-cells but also in human monocytes and macrophages (257, 278), which share the same virus-specific receptors (258), and in B-cell lines (259), in fibroblasts (261) in human brain and the lung (213, 230, 232, 257, 261).
E. No Simian Models for AIDS
Since retroviruses have been isolated from monkeys in captivity with immunodeficiencies and since experimental viremina can depress immune functions in monkeys, such systems are considered to be animal models of human AIDS. For example, 42 of 68 newborn monkeys died with a broad spectrum of diseases that included runting and lymphadenopathy 4 to 6 weeks after inoculation with Mason-Pfizer monkey virus (91). However, this virus has since been found in healthy macaques (262). More recently a retrovirus termed simian AIDS or SAIDS was isolated from monkeys with immunodeficiency (92, 262). Inoculation of three juvenile rhesus monkeys by one isolate was reported to cause splenomegaly and lymphoadenopathy within 2 to 5 weeks. One animal became moribund and two others were alive with simian AIDS at the time of publication (92). However, in another study only transient lymphadenopathy but no lasting AIDS-like disease was observed in macaques inoculated with this virus (263). Another simian virus that is serologically related to AIDS virus, termed STLV-III, was isolated from immunodeficient macaques and from one macaque with a lymphoma. Macaques inoculated with blood or tissue samples of the viral lymphoma died 50 to 60 days later with various diseases (93). However, asymptomatic infections by the same virus have since been identified in no less than 50% of wild green monkeys that did not show any symptoms of a disease (264).
Eight chimpanzees infected with human AIDS virus had not developed symptoms of AIDS 1.5 years past inoculation (265). However, each animal developed antiviral immunity about 1 month after infection, followed by persistent latent infection, as in the human cases (265). A follow-up of chimpanzees inoculated with sera from AIDS patients in 1983 reports no evidence for AIDS in 1986 although the animals had developed antibodies to the virus (243).
Several reasons suggest that these experimental infections of monkeys are not suitable models for human AIDS. Above all, the human virus is not pathogenic in animals. The diseases induced in monkeys by experimental infections with simian viruses all occur fast compared to the 5-year latency for AIDS. Moreover the simian viruses are never associated with a disease in wild animals. Therefore these diseases appear to be exactly analogous to the direct, early pathogenic effects caused by other retroviruses in animals prior to antiviral immunity (see Part I, Section B), and thus are probably models for the early mononucleosis-like diseases which occur in humans infected with AIDS virus prior to antiviral immunity (232, 234, 240) (Section B). Indeed the persistent asymptomatic infections of wild monkeys with simian retroviruses appear to be models for the many asymptomatic infections of humans with AIDS virus or HTLV-I.
F. AIDS Virus as an Indicator of Low Risk for AIDS
The only support for the hypothesis that the AIDS virus causes AIDS is that 90% of the AIDS patients have antibody to the virus. Thus it would appear that the virus, at least as an immunogen, meets the first of Koch's postulates for an etiological agent. This conclusion assumes that all AIDS patients from whom virus cannot be isolated (about 50%) (278) or in whom provirus cannot be demonstrated (85%) and the antibody-negative cases (about 10%) and the virus-free cases reported in one study (3%) (Section C) are false negatives. Indeed the diagnosis of AIDS virus by antibody has recently been questioned on the basis of false positives (234).
At this time the hypothesis that the virus causes AIDS faces several direct challenges. (a) First it fails to explain why active antiviral immunity, which includes neutralizing antibody (225-227) and which effectively prevents virus spread and expression, would not prevent the virus from causing a fatal disease. This is particularly paradoxical since antiviral immunity or "vaccination" typically protects against viral pathogenicity. It is also unexpected that AIDS patients are capable of mounting an apparently highly effective, antiviral immunity, although immunodeficiency is the hallmark of the disease. (b) The hypothesis is also challenged by direct evidence that the virus is not sufficient to cause AIDS. This includes (i) the low percentage of symptomatic infections, (ii) the fact that some infected groups are at a relatively high and others at no risk for AIDS, (iii) the long latent period of the disease (Section B), and (iv) the genetic evidence that the virus lacks a late AIDS function. Since all viral genes are essential for virus replication (28, 245), the virus should kill T-cells and hence cause AIDS at the time of infection rather than 5 years later. (c ) The hypothesis also fails to resolve the contradiction that the AIDS virus, like all retroviruses, depends on mitosis for replication yet is postulated to be directly cytocidal (Section D). (d) The hypothesis offers no convincing explanation for the paradox that a fatal disease would be caused by a virus that is latent and biochemically inactive and that infects less than 1% and is expressed in less than 0.01% of susceptible lymphocytes (Section D). In addition the hypothesis cannot explain why the virus is not pathogenic in asymptomatic infections, since there is no evidence that the virus is more active or further spread in carriers with than in carriers without AIDS.
In view of this it seems likely that AIDS virus is just the most common among the occupational viral infections of AIDS patients and those at risk for AIDS, rather than the cause of AIDS. The disease would then be caused by an as yet unidentified agent which may not even be a virus, since cell-free contacts are not sufficient to transmit the disease. Other viral infections of AIDS patients and those at risk for AIDS include Epstein-Barr and cytomegalovirus in 80 to 90% (222, 268), and herpes virus in 75 to 100%. [D. Purtilo, personal communication.] In addition hepatitis B virus is found in 90% of drug addicts positive for antibody to AIDS virus (267). Among these different viruses, retroviruses are the most likely to be detectable long after infection and hence are the most probable passenger viruses of those exposed to multiple infectious agents. This is because retroviruses are not cytocidal and are unsurpassed in establishing persistent, non-pathogenic infections even in the face of antiviral immunity. Therefore AIDS virus is a useful indicator of contaminated sera that may cause AIDS (13, 27) and that may contain other cell-free and cell-associated infectious agents. It is also for these reasons that latent retroviruses are the most common nonpathogenic passenger viruses of healthy animals and humans. For the same reasons, they are also frequently passenger viruses of slow diseases other than AIDS like the feline, bovine and human leukemias (see Part I) or multiple sclerosis (268) in which latent or defective "leukemia viruses" are occasionally found.
It is concluded that AIDS virus is not sufficient to cause AIDS and that there is no evidence, besides its presence in a latent form, that it is necessary for AIDS. However, the virus may be directly responsible for the early, mononucleosis-like disease observed in several infections prior to antiviral immunity (Section B). In a person who belongs to the high risk group for AIDS, antibody against the AIDS virus serves as an indicator of an annual risk for AIDS that averages 0.3% and may reach 5%, but in a person that does not belong to this group antibody to the virus signals no apparent risk for AIDS. Since nearly all virus carriers have antiviral immunity including neutralizing antibody (225-227), vaccination is not likely to benefit virus carriers with or without AIDS. *
Acknowledgements
I am grateful to R. Cardiff (Davis, CA), K. Cichutek, M. Gardner (Davis, CA), D. Goodrich, E. Humphries (Dallas, TX), J.A. Levy (San Francisco, CA), F. Lilly (New York, NY), G. S. Martin, G. Matioli (Los Angeles, CA), E. Noah (Villingen, Germany), S. Pfaff, W. Phares, D. Purtilo (Omaha, NE), H. Rubin, B. Singer, G. Stent, and R.-P. Zhou for critical comments or review of this manuscript or both and R.C. Gallo (NIH Bethesda, MD) for discussions.
* The abbreviations used are: RSV, Rous sarcoma virus; AIDS, acquired immunodeficiency syndrome; HTLV-1, human T-cell leukemia virus; MMTV, mouse mammary tumor virus; ATLV, adult T-cell leukemia virus; STLV-III, simian T-cell leukemia virus; ATL, adult T-cell leukemia; MCF, mink cell focus-forming; HIV, human immunodeficiency virus; ARV, AIDS-associated retrovirus.
** Koch's postulates define the steps required to establish a microorganism as the cause of a disease: (a) it must be found in all cases of the disease; (b) it must be isolated from the host and grown in pure culture; © it must reproduce the original disease when introduced into a susceptible host; and (d) it must be found present in the experimental host so infected.
Received 6/2/86; revised 10/14/86; accepted 11/11/86.
(This work was) supported by (OIG) National Cancer Institute Grant CA-39915A-01 and Council for Tobacco Research Grant 1547 and by a scholarship in residence of the Fogarty International Center, NIH, Bethesda, MD.
[Exponential power is not printed here, so the phrase "10 (3rd power)" indicates "1,000"]
References
1. Boveri, T. Zur Frage der Entstehung maligner Tumoren. Jena, Germany: Gustav Fischer Verlag, 1914.
2. Foulds, L. Neoplastic Development, Vols. 1 and 2. New York: Academic Press, 1969.
3. Burnett, M.F. The biology of cancer. In: H. German (ed.), Chromosomes and Cancer, Vol. 1, p. 21. New York: John Wiley & Sons, Inc., 1974.
4. Nowell, P.C. The clonal evolution of tumor cell populations. Science (Wash. DC), 194: 23-28, 1976.
5. Duesberg, P.H. Activated proto-onc genes: sufficient or necessary for cancer? Science (Wash.DC), 228: 669-677.
6. Rous, P. The challenge to man of the neoplastic cell. Science (Wash. DC), 157: 24-28, 1967.
7. Stanley, W.M. Virus-induced neoplasia-outlook for the future. Cancer Res., 20: 669-830, 1960.
8. Huebner, R.J., and Todaro, G. Oncogenes of RNA tumors viruses as determinants of cancer. Proc. Natl. Acad. Sci. USA, 64: 1087-1094, 1969.
9. Gross, L. Oncogenic Viruses. Elmsford, NY: Pergamon Press, 1970.
10. Kaplan, H.S. On the natural history of the murine leukemias: Presidential Address. Cancer Res., 27: 1325-1340, 1967.
11. Lwoff, A. Introduction-RNA viruses and host genome in oncogenesis, pp. 1-11. In: P. Emmelot and P. Bentvelzen (eds.), RNA Viruses and Host Genome in Oncogenesis, New York: American Elsevier Co., 1972.
12. Rowe, W.P. Genetic factors in the natural history of murine leukemia virus infection. Cancer Res., 33: 3061-3068, 1973.
13. Weiss, R., Teich, N., Varmus, H., and Coffin, J. RNA Tumor Viruses. Ed. 2. Cold Spring Harbor, NY: Cold Spring Harbor Press, 1985.
14. Tooze, J. The Molecular Biology of Tumor Viruses. Cold Spring Harbor, NY: Cold Spring Harbor Press, 1973.
15. Fenner, F., McAuslan, B.R., Mims, C.A., Sambrook, J., and White, D.O. The Biology of Animal Viruses. New York: Academic Press, 1974.
16. Weiss, R., Teich, N., Varmus, H., and Coffin, J. RNA Tumor Viruses. Ed. 2. Cold Spring Harbor, NY: Cold Spring Harbor Press, 1982.
17. Levy, J.A. Xenotropic type C viruses. Curr. Top. Microbiol. Immunol., 79: 111-123, 1978.
18. Kozak, C., and Silver, J. The transmission and activation of endogenous mouse retroviral genomes. TIG Rev., 1: 331-334, 1985.
19. Duesberg, P.H. Retroviral Transforming genes in normal cells? Nature (Lond.), 304: 219-225, 1983.
20. Duesberg, P.H. Transforming genes of retroviruses. Cold Spring Harbor Symp. Quant. Biol., 44: 13-29, 1980.
21. Duesberg, P.H., and Vogt, P.K. Differences between the ribonucleic acid of transforming and nontransforming avian tumor viruses. Proc. Natl. Acad. Sci. USA, 67: 1673-1680, 1970.
22. Duesberg, P.H., Vogt, P.K., Beemon, K., and Lai, M. Avian RNA tumor viruses: mechanism of recombination and complexity of the genome. Cold Spring Harbor Symp. Quant. Biol., 39: 847-857, 1974.
23. Wang, L.H., Duesberg, P.H., Beemon, K., and Vogt, P.K. Mapping RNase T-1-resistant oligonucleotides of avian tumor virus RNAs: sarcoma-specific oligonucleotides are near the poly(A) end and oligonucleotides common to sarcoma and transformation-defective viruses are at the poly(A) end. J. Virol., 16: 1051-1070, 1975.
24. Wang, L.H., Galehouse, D., Mellon, P., Duesberg, P., Mason, W.S., and Vogt, P.K. Mapping oligonucleotides of Rous sarcoma virus RNA that segregate with polymerase and group-specific antigen markers in recombinants. Proc. Natl. Acad. Sci. USA, 73: 3952-3956, 1976.
25. Rubin, H., and Temlin, H.M. A radiological study of cell-virus interaction in the Rous sarcoma. Virology, 7: 75-91, 1958.
26. Barre-Sinoussi, F., Chermann, J.C., Rey, F., Nugeyre, M.T., Chamaret, S., Gruest, J., Danguet, C., Axler-Blin, C., Vezinet-Brun, F., Rouzioux, C., Rosenbaum, W., and Montagnier, L. Isolation of T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science (Wash. DC), 220: 868-870, 1983.
27. Wong-Staal, F., and Gallo, R.C. Human T-lymphotropic retroviruses. Nature (Lond.), 317: 395-403, 1985.
28. Sodroski, J., Goh, W.C., Rosen, C., Dayton, H., Terwilliger, E., and Haseltine, W. A second post-transcriptional trans-activator gene required for HTLV-III replication. Nature (Lond.), 321: 412-417, 1986.
29. Ellermann, V., and Bang, O. Experimentelle leukaemie bei Huehnern. Centralblatt Bakteriol. Parasitenk. Abt. I Orig., 46: 595-609, 1908.
30. Burmester, B.R., Gentry, R.F., and Waters, N.F. The presence of the virus of visceral lymphomatosis in embrionated eggs of normal appearing hens. Science (Wash. DC), 34: 609-617, 1955.
31. Rubin, H., Fanshier, L., Cornelius, A., and Hughes, W.F. Tolerance and immunity in chickens after congenital and contact infection with avian leukosis virus. Virology, 17: 143-156, 1962.
32. Purchase, H.G., and Burmester, B.R. Leukosis sarcoma group. In: M.S. Hofstad et al. (eds.), Diseases of Poultry, Ed. 6, pp. 502-568. Ames, IA: The Iowa State University Press, 1972.
33. Calnek, B.W. Neoplastic diseases. Introduction. In: M.S. Hofstad et al. (eds.), Diseases of Poultry, Ed. 6, pp. 467-470. Ames, IA: The Iowa State University Press, 1972.
34. Calnek, B.W., and Witter, R.L. Marek's disease. In: M.S. Hofstad et al. (eds.), Diseases of Poultry, Ed. 6, pp. 470-502. Ames, IA: The Iowa State University Press, 1972.
35. Baba, T.W., and Humphries, E.H. Avian leukosis virus infection: analysis of viremia and DNA integration in susceptible and resistant chicken lines. J. Virol., 51: 123-130, 1984.
36. Baba, T., and Humphries, E.H. Formation of a transformed follicle is necessary but not sufficient for development of an avian leukosis virus-induced lymphoma. Proc. Natl. Acad. Sci. USA, 82: 213-216, 1985.
37. Neiman, P., Payne, L.N., and Weiss, R.A. Viral DNA in bursal lymphomas induced by avian leukosis viruses. J. Virol., 34: 178-186, 1980.
38. Gardner, M.B. Type C viruses and wild mice: characterization and natural history of amphotropic, ecotropic, and xenotropic MuLV. Curr. Top. Microbiol. Immunol., 79: 215-259, 1978.
39. Gardner, M.B., Rasheed, S., Estes, J.D., Casagrande, J., Ihle, J.N., and Stephenson, J.R. The history of viruses and cancer in wild mice. Cold Spring Harbor Conf. Cell Proliferation, 7: 971-987, 1980.
40. Lilly, F., Duran-Reynals, M.L., and Rowe, W.P. Correlation of early murine leukemia virus titer and H-2 type of spontaneous leukemia in mice of the BALB X AKR cross: a genetic analysis. J. Exp. Med., 141: 882-889, 1975.
41. Melamedoff, M., Lilly, F., and Duran-Reynals, M.L. Suppression of endogenous murine leukemia virus by maternal resistance factors. J. Exp. Med., 158: 506-514, 1983.
42. Lee, J.C., and Ihle, J.N. Chronic immune stimulation is required for Moloney leukemia virus-induced lymphomas. Nature (Lond.), 289: 407-409, 1981.
43. Pluznik, D.H., and Sachs, L. Quantitation of a murine leukemia virus with a spleen colony assay. J. Natl. Cancer Inst., 33: 535-546, 1964.
44. Duesberg, P.H., and Robinson, W.S. Nucleic acid and proteins isolated from the Rauscher mouse leukemia virus (MLV). Proc. Natl. Acad. Sci. USA, 55: 219-227, 1966.
45. Klement, V., and Nicolson, M.O. Methods for assays of RNA tumor viruses. Methods Virol., 6: 60-108, 1977.
46. Ihle, J.N., and Lee, I.C. Possible immunological mechanisms in C-type viral leukemogenesis in mice. Curr. Top. Microbiol. Immunol., 98: 85-100, 1981.
47. Mosier, D.E., Yetter, R.A., and Morse, H.C. Retrovirus induction of acute lymphoproliferative disease and profound immunosuppression in adult C57BL/6 mice. J. Exp. Med. 161: 766-784, 1985.
48. Rowe, W.P., and Pincus, T. Quantitative studies of naturally occurring murine leukemia virus infection of AKR mice. J. Exp. Med., 135: 429-436, 1972.
49. Crittenden, L.B. New hypotheses for viral unduction of lymphoid leukosis in chickens. Cold Spring Harbor Conf. Cell Proliferation, 7: 529-541, 1980.
50. Robinson, H.L., and Gagnon, G.C. Patterns of proviral insertion and deletion in avian leukosis virus-induced lymphomas. J. Virol., 57: 28-36, 1986.
51. Little, C.C., per staff of Roscoe B. Jackson Memorial Laboratory. The existence of non-chromosomal influence in the incidence of mammary tumors in mice. Science (Wash. DC), 78: 465-466, 1933.
52. Bittner, J.J. The milk-influence of breast tumors in mice. Science (Wash. DC), 95: 462-463, 1942.
53. Cohen, J.C., and Varmus, H.E. Endogenous mammary tumor virus DNA varies among wild mice and segregates during inbreeding. Nature (Lond.), 278: 418-423, 1979.
54. Cardiff, R.D., and Young, L.J.T. Mouse mammary tumor biology: a new synthesis. Cold Spring Harbor Conf. Cell Proliferation, 7: 1105-1114, 1980.
55. Bentvelzen, P., Brinkhof, J., and Westenbrink, F. Expression of endogenous mammary tumor virus in mice: its genetic control and relevance to spontaneous mammary carcinogenesis. Cold Spring Harbor Conf. Cell Proliferation, 7: 1095-1104, 1980.
56. Cardiff, R.D., Morris, D.W., Young, L.J.T., and Strange, R. MuMTV genotype, proto-neoplasia, and tumor progression. In: M.A. Rich, J.C. Hager, and J. Taylor-Papadimitriou (eds.), Breast Cancer Origins, Detection and Treatment. Proceedings of the International Breast Cancer Research Conference, London, March 24-28, pp. 156-166. The Hague: Martinus Nijhoff Publishing, 1985.
57. Pathak, V.K., Strange, R., Young, L.J.T., Morris, D.W., and Cardiff, R.D. A survey of Int Region DNA rearrangements in C3H and BALB/cfC3H mouse mammary tumor systems. J. Natl. Cancer Inst., in press, 1987.
58. Altrock, B.W., and Cardiff, R.D. Mouse mammary tumor virus infection: virus expression and tumor risk potential. J. Nat'l. Cancer Inst., 63: 1075-1080, 1979.
59. Jarrett, W.F.H., Crawford, A.M., Martin, W.B., and Davie, F. Leukemia in the cat: a virus-like particle associated with leukemia (lymphosarcoma). Nature (Lond.), 202: 566-568, 1964.
60. Roberson, P., Jarrett, W., and Mackey, L. Epidemiological studies of feline leukemia virus infection I. A serological survey in urban cats. Int. J. Cancer, 15: 781-785, 1985.
61. Essex, M. Horizontally and vertically transmitted oncorna-viruses of cats. Adv. Cancer Res., 21: 175-248, 1975.
62. Dorn, C.R., Taylor, D.O.N., Schneider, R., Hibbard, H.H., and Klauber, M.R. Survey of animal neoplasms in Alameda and Contra Costa counties, California. II. Cancer morbidity in dogs and cats from Alameda County. J. Nat'l. Cancer Inst., 40: 307-318, 1968.
63. Essex, M. Feline leukemia and sarcoma viruses. In: G. Klein (ed.), Viral Oncology, pp. 205-229, New York: Raven Press, 1960.
64. Jarrett, W.F., Jarrett, O., Mackey, L., Laird, W., Hardy, W.D., and Essex, M. Horizontal transmission of leukemia virus and leukemia in the cat. J. Nat'l. Cancer Inst., 51: 833-841, 1973.
65. Rojko, J.S., Hoover, E.A., Quackenbush, S.L., and Olsen, R.G. Reactivation of latent feline leukemia virus infection. Nature (Lond.), 298: 385-388, 1972.
66. Gutensohn, N., Essex, M., Francis, D.P., and Hardy, W.D. Risk of humans from exposure to feline leukemia virus: epidemiological considerations. Cold Spring Harbor Conf. Cell Proliferation, 7: 699-706, 1980.
67. Anderson, L.J., Jarrett, W.F.M., Jarrett, O., and Laird, H.M. Feline leukemia-virus infection of kittens: mortality associated with atrophy of the thymus and lymphoid depletion. J. Nat'l. Cancer Inst., 47: 807-817, 1971.
68. Hardy, W.D., Old, L.J., Hess, P.W., Essex, M., and Cotter, S. Horizontal transmission of feline leukemia virus. Nat. New Biol., 244: 266-269, 1973.
69. Kawatami, T.G., Sun, L., and McDowell, F.S. Infectious primate type-C virus shed by healthy gibbons. Nature (Lond.), 268: 448-450, 1977.
70. Kawatami, T.G., and McDowell, F.S. Factors regulating the onset of chronic myelogenous leukemia in gibbons. Cold Spring Harbor Conf. Cell Proliferation, 7: 719-727, 1980.
71. Poiesz, B.J., Ruscetti, F.W., Gazdar, A.F., Bunn, P.A., Minna, J.D., and Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Porc. Nat'l. Acad. Sci. USA, 77: 7415-7419, 1980.
72. Miller, J.M., Miller, L.D., Olson, C., and Gillette, K.G. Virus-like particles in phytohemagglutinin stimulated lymphocyte cultures with reference to bovine lymphosarcoma. J. Nat'l. Cancer Inst., 43: 1297-1305, 1969.
73. Miller, J.M., and van der Maaten, M.S. The biology of bovine leukemia virus infection in cattle. Cold Spring Harbor Conf. Cell Proliferation, 7: 901-999, 1980.
74. Mussgay, M., Dietzschold, R., Lorenz, R., Matheka, H.-D., Mattheus, W., Straub, O.C., Welland, F., Wilesmith, J.W., Frenzel, B., and Kaaden, O.R. Some properties of bovine leukemia virus, its use in seroepidemiological studies and eradication of the disease from infected herds. Cold Spring Harbor Conf. Cell Proliferation, 7: 911-925, 1980.
75. Kenyon, S.Y., Ferrer, J.F., McFeely, R.A., and Graves, D.C. Induction of lymphosarcoma in sheep by bovine leukemia virus. J. Nat'l. Cancer Inst., 67: 1157-1163, 1981.
76. Hoxie, J.E., Matthews, D.M., and Ames, D.B. Infection of human endothelial cells by human T-cell virus. Proc. Nat. Acad. Sci. USA, 81: 7591-7595, 1984.
77. Yoshikura, H., Nishida, J., Yoshida, M., Kitamura, Y., Takaku, F., and Ikeda, S. Isolation of HTLV derived from Japanese adult T-cell leukemia patients in human diploid fibroblast strain IMR90 and the biological characters of the infected cells. Int. J. Cancer, 33: 745-749, 1984.
78. Tajima, K., and Kuroishi, T. Estimation of rate of incidence of ATL among ATLV (HTLV-I) carriers in Kyushu, Japan. Jpn. J. Clin. Oncol., 15: 423-430, 1985.
79. Maeda, Y., Furakawu, M., Takehara, Y., Yoshimura, Y., Miyumoto, K., Matsubara, T., Morishima, Y., Tajima, K., Okochi, K., and Hinuma, Y. Prevalence of possible adult T-cell leukemia virus-carriers among volunteer blood donors in Japan: a nation-wide study. Int. J. Cancer, 33: 717-730, 1984.
80. Pan, I.-H., Chung, C.-S., Komoda, H., Mai, J., and Hinuma, Y. Seroepidemiology of adult T-cell leukemia virus in Taiwan. Jpn. J. Cancer Res., 76: 9-11, 1985.
81. Okochi, K., Sato, H., and Hinuma, Y. A retrospective study on transmission of adult T-cell leukemia virus by blood transfusion: seroconversion of recipients. Vox Sang., 46: 245-253, 1984.
82. Kaplan, H.S. Mouse leukemia: interaction of multiple components in a complex oncogenic system. In: P. Emmelot and P. Bentvelzen (eds.), RNA Viruses and Host Genome in Onco-genesis, pp. 143-154. New York: North Holland, American Elsevier, 1972.
83. Fenner, F., and White, D.O. Medical Virology. New York: Academic Press, 1970.
84. Carter, J.K., Procter, F.J., and Smith, R.E. Induction of angiosarcomas by ring-necked pheasant virus. Infect. Immun., 40: 310-319, 1983.
85. Carter, J.K., and Smith, R.E. Rapid induction of hyperthyroidism by an avian leukosis virus. Infect. Immun., 40: 795-805, 1983.
86. Smith, R.E., and Morgan, J.H. Identification of plaque isolates of an avian retrovirus causing rapid and slow onset osteopetrosis. Virology, 119: 488-499, 1982.
87. Morgan, J.H., and Smith, R.E. Rapid induction of osteopetrosis by subgroup E. recombinant viruses. Virology 129: 493-500, 1983.
88. Carter, J.K., Ow, C.L., and Smith, R.E. Rous-associated virus type 7 induces a syndrome in chickens characterized by stunting and obesity. Infect. Immun., 39: 410-422, 1983.
89. Smith, R.E., and Van Eldik, J. Characterization of the immune suppression accompanying virus-induced avian osteopetrosis. Infect. Immun., 22: 452-461, 1978.
90. Rojko, J.S., Hoover, E.A., Lawrence, E.M., Richard, G.O., and Schaller, J.P. Pathogenesis of Experimental Feline Leukemia Virus Infection. J. Nat'l. Cancer Inst., 63: 759-765, 1979.
91. Fine, D.L., Landon, J.C., Pienta, R.J., Kubicek, M.T., Valerio, M.G., Leob, W.F., and Chopra, H.C. Responses of infant rhesus monkeys to inoculation with Mason-Pfizer monkey virus materials. J. Nat'l. Cancer Inst., 54: 651-658, 1975.
92. Marx, P.A., Maul, D.H., Osborn, K.G., Lerce, N.W., Moody, P., Lowenstine, L.J., Henrickson, R.V., Arthur, L.O., Gravell, M., London, W.T., Lever, J.L., Levy, J.A., Munn, P., and Gardner, M.B. Simian AIDS: isolation of a type D retrovirus and disease transmission. Science (Wash. DC), 223: 1083-1086, 1984.
93. Daniel, M.D., Letvin, N.L., King, N.W., Kannagi, M., Sehgal, P.K., Hunt, R.D., Kanki, P.J., Essex, M., and Desrosiers, R.C. Isolation of T-cell tropic HTLV-III-like retrovirus from macaques. Science (Wash. DC), 228: 1201-1204, 1985.
94. Breindel, M., Harbers, K., and Jaenisch, R. Retrovirus-induced lethal mutation in collagen I gene of mice is associated with an altered chromatin structure. Cell, 38: 9-16, 1984.
95. King, W., Patel, M.D., Lobel, L.I., Goff, S.P., and Nguyen-Huu, M.C. Insertion mutagenesis of embryonal carcinoma cells by retroviruses. Science (Wash. DC), 228: 554-558, 1985.
96. Purchase, H.G., Ludford, C., Nazerian, K., and Cox, H.W. A new group of oncogenic viruses: reticuloendotheliosis, chick syncytial, chick infectious anemia, and spleen necrosis virus. J. Nat'l. Cancer Inst., 51: 489-499, 1973.
97. Keshet, E., and Temin, H.M. Cell killing by spleen necrosis virus is correlated with a transient accumulation of spleen necrosis virus DNA. J. Virol., 31: 376-388, 1979.
98. Graf, T. A plaque assay for avian RNA tumor viruses. Virology, 50: 567-578, 1972.
99. Klement, V., Rowe, W.P., Hartley, J.W., and Pugh, W.E. Mixed culture cytopathogenicity: a new test for growth of murine leukemia viruses in tissue culture. Proc. Nat'l. Acad. Sci. USA, 63: 753-758, 1969.
100. Hartley, J.W., Wolford, N.K., Old, L.J., and Rowe, W.P. A new class of murine leukemia virus associated with development of spontaneous lymphomas. Proc. Nat'l. Acad. Sci. USA, 74:
789-792, 1977.
101. Rangan, S.R.S., Ueberhorst, P.J., and Wong, M.C. Syncytial giant cell focus assay for viruses derived from feline leukemia and a simian sarcoma. Proc. Soc. Exp. Biol. Med., 142: 1077-1082, 1973.
102. Rangan, S.R.S., Wong, M.C., Ueberhorst, P.J., and Ablashi, D.J. Mixed culture cytopathogenecity induced by virus preparations derived from simian sarcoma virus (SSV-1) infected cultures. J. Nat'l. Cancer Inst., 49: 571-577, 1972.
103. Rand, K.H., Long, C.W., Wei, T.T., and Gilden, R.V. Plaque assay for the Mason-Pfizer monkey virus. J. Nat'l. Cancer Inst., 53: 449-452, 1974.
104. Diglio, C.A., and Ferrer, J.P. Induction of syncytia by the bovine C-type leukemia virus. Cancer Res., 36: 1056-1057, 1976.
105. Nagy, K., Clapham, P., Cheingsong-Popov, R., and Weiss, R.A. Human T-cell leukemia virus type I: induction of syncytia and inhibition by patient's sera. Int. J. Cancer, 32: 321-328, 1983.
106. Hoshino, H., Shinoyama, M., Miwa, M., and Sugimura, T. Detection of lymphocytes producing a human retrovirus associated with adult T-cell leukemia by syncytia induction assay. Proc. Nat'l. Acad. Sci. USA, 80: 7337-7341, 1983.
107. Baba, T.W., and Humphries, E.H. Differential response to avian leukosis virus infection exhibited by two chicken lines. Virology, 135: 181-188, 1985.
108. Rich, M.A. Virus-induced murine leukemia. In: M.A. Rich (ed.), Experimental Leukemia, pp. 15-49. New York: Appleton-Century-Crofts, 1968.
109. Bentvelzen, P. The putative mam gene of the murine mammary tumor virus. In: G. Klein (ed.), Advances in Viral Oncology, pp. 141-152. New York: Raven Press, 1982.
110. Hardy, W.D., McClelland, A.J., Zuckerman, E.E., Snyder, H.W., MacEwen, E.G., Francis, D.P., and Essex, M. Immunology and epidemiology of feline leukemia virus nonproducer lympho-sarcomas. Cold Spring Harbor Cancer Conf. Cell Proliferation, 7: 677-697, 1980.
111. Kinoshita, K., Hino, S., Amagasaki, T., Yamada, Y., Kamihira, E., Ichimura, M., Munehisa, T., and Hinuma, Y. Development of adult T-cell leukemia-lymphoma (ATL) in two anti-ATL-associated antigen-positive healthy adults. Gann, 73: 684-685, 1982.
112. Kinoshita, K., Amagasaki, T., Yamada, Y., Kamihira, E., Ichimura, M., Kamihira, S., Yamada, Y., Momita, S., Kusano, M., Morikawa, T., Fujita, S., Ueda, Y., Ito, N., and Yoshida, M. Preleukemic state of adult T cell leukemia: abnormal T lympho-cytosis induced by human adult T cell leukemia-lymphoma virus. Blood, 66: 120-127, 1985.
113. Gallo, R.C., Wong-Staal, F., Markham, P.D., Ruscetti, F., Kalyanaraman, V.S., Ceccherini-Nelli, L., Dalla Favera, R., Josephs, S., Miller, N.R., and Reitz, M.S. Recent studies with infectious primate retroviruses: hybridization to primate DNA and some biological effects on fresh human blood leukocytes by simian sarcoma virus and gibbon ape leukemia virus. Cold Spring Harbor Conf. Cell Proliferation, 7: 753-773, 1980.
114. Miyoshi, I., Yoshimoto, S., Kubonishi, I., Taguchi, H., Shiraishi, Y., Oktsuki, Y., and Akagi, T. Transformation of normal human cord lymphocytes by co-cultivation with a lethally irradiated human T-cell line carrying type C virus particles. Gann, 72: 997-998, 1981.
115. DeRossi, A., Aldovini, A., Franchini, G., Mann, D., Gallo, R.C., and Wong-Staal, F. Clonal selection of T lymphocytes infected by cell-free human T-cell leukemia/lymphovirus type I: parameters of virus integration and expression. Virology, 143: 640-645, 1985.
116. Nowell, P.C., Finan, J.B., Clark, J.W., Sarin, P.S., and Gallo, R.C. Karyotypic differences between primary cultures and cell lines from tumors with the human T-cell leukemia virus. J. Nat'l. Cancer Inst., 73: 849-852, 1984.
117. Crittenden, L.B. Two levels of genetic resistance to lymphoid leukosis. Avian Dis., 19: 281-292, 1975.
118. Fung, Y.-K.T., Fadly, A.M., Crittenden, L.B., and Kung, H.-J. Avian lymphoid leukosis virus infection and DNA integration in the preleukotic bursal tissues: a comparative study of susceptible and resistant lines. Virology, 119: 411-421, 1982.
119. Raines, M.A., Lewis, W.G., Crittenden, L.B., and Kung, H.-J. c-erb B activation in avian leukosis virus-induced erythroblastosis: clustered integration sites and the arrangement of provirus in the c-erb B alleles. Proc. Nat'l. Acad. Sci. USA, 82: 2287-2291, 1985.
120. Robinson, H.L., Miles, B.D., Catalano, D.E., Broles, W.E., and Crittenden, L.B. Susceptibility to erb B-induced erythroblastosis is a dominant trait of 15I chickens. J. Virol., 55: 617-622, 1985.
121. Crittenden, L.B., Witter, R.L., Okazaki, W., and Neiman, P.E. Lymphoid neoplasms in chicken flocks free of infection with exogenous avian tumor viruses. J. Nat'l. Cancer Inst., 63: 191-200, 1979.
122. Miles, B.D., and Robinson, H.L. High-frequency transduction of c-erb B in avian leukosis virus-induced erythroblastosis. J. Virol., 54: 295-303, 1985.
123. Purchase, H.G., Gilmour, P.G., Romero, C.H., and Okazaki, W. Post-infection genetic resistance to avian lymphoid leukosis resides in B target cells. Nature (Lond.), 270: 61-62, 1977.
124. Bacon, L.D., Witter, R.L., Crittenden, L.B., Fadly, A., and Motta, J. B-haplotype influence on Marek's disease, Rous sarcoma, and lymphoid leukosis virus-induced tumors in chickens. Poult. Sci., 60: 1132-1139, 1981.
125. Meruelo, D., Nimelstein, S.H., Jones, P.P., Lieberman, M., and McDevitt, H.O. Increased synthesis and expression of H-2 antigens on thymocytes as a result of leukemia virus infection: a possible mechanism for H-2 linked control of virus-induced neoplasia. J. Exp. Med., 147: 470-487, 1978.
126. Evans, L.H., Duesberg, P.H., and Scolnick, E.M. Replication of spleen focus-forming Friend virus in fibroblasts form C57BL mice that are genetically resistant to spleen focus formation. Virology, 101: 534-539, 1980.
127. Dux, A. Genetic aspects in the genesis of mammary cancer. In: P. Heston, W.E. Genetic factors in tumorigenesis. In: P. Emmelot and P. Bentvelzen (eds.), RNA Viruses and Host Genome in Oncogenesis, pp. 301-308. New York: American Elsevier, 1972.
128. Heston, W.E. Genetic factors in tumorigenesis. In: P. Emmelot and P. Bentvelzen (eds.), RNA Viruses and Host Genome in Oncogenesis, pp. 13-24. New York: American Elsevier, 1972.
129. Moore, D.H., Long, C.A., Vaidya, A.B., Sheffield, J.B., Dion, A.G., and Lasfargues, E.Y. Mammary tumor viruses. Adv. Cancer Res., 29: 347-418, 1979.
130. Hayward, W.S., Neel, B.G., and Astrin, S.M. Activation of a cellular onc gene by promoter insertions in ALV-induced lymphoid leukosis. Nature (Lond.), 290: 475-480, 1981.
131. Neel, G.B., Hayward, W.S., Robinson, H.L., Fong, J., and Astrin, S.M. Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: oncogenesis by promoter insertion. Cell, 23: 323-324, 1981.
132. Payne, G.S., Bishop, J.M., and Varmus, H.E. Multiple arrangements of viral DNA and an activated host oncogene in bursal lymphoma. Nature (Lond.), 295: 209-214, 1982.
133. Astrin, S.M., Robinson, H.L., Crittenden, L.B., Buss, E.G., Wyban, J., and Hayward, W.S. Ten genetic loci in the chicken that contain structural genes for endogenous avian leukosis virus. Cold Spring Harbor Symp. Quant. Biol., 44: 1105-1109, 1980.
134. Weiss, R.A., and Frisby, D.P. Are avian endogenous viruses pathogenic? In: D.J. Yohn and J.R. Blakeslee (eds.), Advances in Comparative Leukemia Research 1981, pp. 303-311. Amsterdam: Elsevier/North-Holland Biomedical Press, Inc., 1982.
135. Horak, I., Lee, J.C., Enjuanes, L., and Ihle, J.N. Characterization of a unique defective type C-virus associated with a Moloney leukemia virus-induced splenic T-cell lymphoma cell line. J. Virol., 30: 299-308, 1980.
136. Corcoran, L.M., Adams, J.M., Dunn, A.R., and Cong. S. Murine T lymphomas in which the cellular myc oncogene has been activated by retroviral insertion. Cell, 37: 113-122, 1984.
137. Tsichlis, P.N., Strauss, P.G., and Kozak, C.A. Cellular DNA region involved in induction of thymic lymphomas (Mlvi-2) maps to mouse chromosome 15. Mol. Cell Biol., 4: 997-1000, 1984.
138. McGrath, C.M., Nandi, S., and Young, L.J.T. Relationship between organization of mammary tumor and the ability of tumor cells to replicate mammary tumor virus and to recognize growth-inhibitory contact signals in vitro. J. Virol., 9: 367-376, 1972.
139. Bentvelzen, P. Hereditary infections with mammary tumor viruses in mice. In: P. Emmelot and P. Bentvelzen (eds.), RNA Viruses and Host Genome in Oncogenesis, pp. 309-337. New York: American Elsevier, 1972.
140. Peters, G., Brookes, S., Smith, R., and Dickson, C. Tumorigenesis by mouse mammary tumor virus: evidence for a common region for provirus integration in mammary tumors. Cell, 33: 369-377, 1983.
141. Gardner, M.B., Malone, R.W., Morris, D.W., Young, L.J.T., Strange, R., Cardiff, R.D., Foulkin, L.J., and Mitchell, D.J. Mammary tumors in feral mice lacking MuMTV DNA. J. Exp. Pathol., 2: 93-98, 1985.
142. Michalides, R., Van Deemter, L., Nusse, R., and Hegeman, P.C. Induction of mouse mammary tumor virus RNA in mammary tumors of BALB/c mice treated with urethane, X-irradiation and hormones. J. Virol., 31: 63-72, 1979.
143. Levy, L.S., Gardner, M.B., and Casey, J.W. Isolation of a feline leukaemia provirus containing the oncogene myc from a feline lymphosarcoma. Nature (Lond.), 308: 853-856, 1984.
144. Mullins, J.L., Brody, D.S., Binasi, R.C., and Cotten, S.M. Viral transduction of c-myc in naturally occurring feline leukemias. Nature (Lond.), 308: 856-858, 1984.
145. Neil, J.C., Hughes, D., McFarlane, R., Wilkie, N.M., Onions, D.E., Lees, G., and Jarrett, O. Transduction and rearrangement of the myc gene by feline leukaemia virus in naturally occurring T-cell leukaemias. Nature (Lond.), 308: 814-820, 1984.
146. Kettmann, R., Deschamps, J., Clerter, Y., Couez, D., Burny, A., and Marbaix, G. Leukemogenesis by bovine leukemia virus: Proviral DNA integration and lack of RNA expression of viral long terminal repeat and 3' proximate cellular sequences. Proc. Nat'l. Acad. Sci. USA, 79: 2465-2469, 1982.
147. Rosen, C.A., Sodroski, J.G., Kettmann, R., Burny, A., and Hazeltine, W.A. trans activation of the bovine leukemia virus long terminal repeat in BVL-infected cells. Science (Wash. DC), 227: 320-322, 1985.
148. Kettmann, R., Deschamps, J., Couez, D., Claustriaux, J.J., Palm, R., and Burny, A. Chromosome integration domain for bovine leukemia provirus in tumors. J. Virol., 47: 146-150, 1983.
149. Hoshino, H., Esumi, H., Miwa, M., Shimoyama, M., Minato, K., Tobinai, K., Hirose, M., Natunabe, S., Inadu, N., Konishita, K., Komihira, S., Ichimara, M., and Sugimara, T. Establishment and characterization of 10 cell lines derived from patients with adult T-cell leukemia. Proc. Nat'l. Acad. Sci. USA, 80: 6061-6065, 1983.
150. Franchini, G., Wong-Staal, F., and Gallo, R.C. Human T-cell leukemia virus (HTLV-I) transcripts in fresh and cultured cells of patients with adult T-cell leukemia. Proc. Nat'l. Acad. Sci. USA, 81: 6207-6211, 1984.
151. Wong-Staal, F., Franchini, G., Hahn, B., Arya, S., Gelmann, E.P., Manzari, V., and Gallo, R.C. Human T-cell leukemia/lymphoma virus, in vitro transformation, and leukemia: some molecular studies. In: R.C. Gallo, M. Essex, and L. Gross (eds.), Human T-cell Leukemia-Lymphoma, pp. 133-139. Cold Spring Harbor, NY: Cold Spring Harbor Press, 1984.
152. Sodroski, J.G., Rosen, C.A., and Haseltine, W.A. trans-Acting transcriptional activation of the long terminal repeat of human T lymphotropic viruses in infected cells. Science (Wash. DC), 225: 381-385, 1984.
153. Burny, A. More and better transactivation. Nature (Lond.), 321: 378, 1986.
154. Yoshida, M., Seiki, M., Hattori, S., and Watanabe, F. Genome structure of human T-cell leukemia virus and its involvement in the development of adult T-cell leukemia/lymphoma. In: R.C. Gallo, M. Essex, and L. Gross (eds.), Human T-cell Leukemia-Lymphoma Virus, pp. 141-149. Cold Spring Harbor, NY: Cold Spring Harbor Press, 1984.
155. Yoshida, M., Seiki, M., Yamaguchi, Y., and Takatsuki, K. Monoclonal integration of HTLV in all primary tumors of adult T-cell leukemias suggests causative role of HTLV in the disease. Proc. Nat'l. Acad. Sci. USA, 81: 2534-2537, 1984.
156. Takenaka, T., Nakamine, H., Oshiro, I., Maeda, J., Kobori, M., Hayashi, T., Komoda, H., Hinuma, Y., and Hanaoka, M. Serologic and epidemiologic studies on adult T-cell leukemia (ATL): special reference to a comparison between patients with and without antibody to ATL-associated antigen. Jpn. J. Clin. Oncol., 13(Suppl. 2): 257-268, 1983.
157. Tobinai, K., Nagai, M., Setoya, T., Shibata, T., Minato, K., and Shimoyama, M. Anti-ATLA (antibody to adult T-cell leukemia virus-associated antigen), highly positive in OKT4-positive mature T-cell malignancies. Jpn. J. Clin. Oncol., 13(Suppl. 2): 237-244, 1983.
158. Shimoyama, M., Kagami, Y., Shimotohno, K., Miwa, M., Minato, K., Tobinai, K., Suemasu, K., and Sugimura, T. Adult T-cell leukemia/lymphoma not associated with human T-cell leukemia virus type I. Proc. Nat'l. Acad. Sci. USA, 83: 4524-4528, 1986.
159. Manzari, V., Gradilone, A. Basillari, G., Fani, M., Collalti, E., Pandolfi, F., DeRossi, G., Liso, V., Babbo, P., Robert-Guroff, M., and Frati, L. HTLV-I is endemic in southern Italy: detection of the first infectious cluster in a white population. Int. J. Cancer, 36: 557-559, 1985.
160. Pandolfi, F., De Rossi, G., Lauria, F., Ranucci, A., Barillari, G., Manzari, V., Semenzato, G., Liso, V., Pizzolo, G., and Aiuti, F. T-helper phenotype chronic lymphocytic leukeamia and "adult T-cell leukemia" in Italy. Lancet, 2: 633-636, 1985.
161. Nagy, K., Gyuris, A., Drexler, E., Foldes, I., Fust, G., and Horvath, A. Human T-lymphotropic virus infections in Hungary. In: Proceedings of the 14th International Cancer Congress Budapest, August 21-27, Abstract 1861, 1986.
162. Bishop, J.M. Enemies within the genesis of retrovirus oncogenes. Cell, 23: 5-6, 1981.
163. Steffen, D., and Weinberg, R.A. The integrated genome of murine leukemia virus. Cell, 15: 1003-1010, 1978.
164. Canaani, E., and Aaronson, S.A. Restriction enzyme analysis of mouse cellular type C viral DNA: emergence of new viral sequences in spontaneous AKR lymphomas. Proc. Nat'l. Acad. Sci. USA, 76: 1677-1687, 1979.
165. Seiki, M., Eddy, R., Shows, T.B., and Yoshida, M. Nonspecific integration of the HTLV provirus genome into adult T-cell leukaemia cells. Nature (Lond.), 309: 640-642, 1984.
166. Hann, S.R., Abrams, H., Rohrschneider, L., and Eisenman, R. Proteins encoded by v-myc and c-myc oncogenes: identification and localization in acute leukemia virus transformants and bursal lymphoma cell lines. Cell, 34: 789-798, 1983.
167. Shih, C.-K., Linial, M., Goodenow, M.M., and Hayward, W.S. Nucleotide sequence 5' of the chicken c-myc coding region: localization of a non-coding exon that is absent from myc transcripts in most avian leukosis virus-induced lymphomas. Proc. Nat'l. Acad. Sci. USA, 81: 4697-4701, 1984.
168. Pachel, C., Schubach, W., Eisenman, R., and Linial, M. Expression of c-myc RNA in bursal lymphoma cell lines: identification of c-myc encoded proteins by hybrid-selected translation. Cell, 33: 335-344, 1983.
169. Schubach, W., and Groudine, M. Alteration of c-myc chromatin structure by avian leukosis virus integration. Nature (Lond.), 307: 702-708, 1984.
170. Li, Y., Holland, C., Hartley, J., and Hopkins, N. Viral integration near c-myc in 10-20% of MCF247-induced AKR lymphomas. Proc. Nat'l. Acad. Sci. USA, 81: 6808-6811, 1984.
171. Steffen, D. Proviruses are adjacent to c-myc in some murine leukemia virus-induced lymphomas. Proc. Nat'l. Acad. Sci. USA, 81: 2097-2101, 1984.
172. O'Donnel, P.V., Fleissner, E., Lonial, H., Koehne, C.F., and Reicin, A. Early clonality and high-frequency proviral integration into the c-myc locus in AKR leukemias. J. Virol., 44: 500-503, 1985.
173. Nusse, R., and Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell, 31: 91-109, 1982.
174. Peters, G., Kozak, C., and Dickson, C. Mouse mammary tumor virus integration regions int-1 and int-2 map on different mouse chromosomes. Mol. Cell Biol., 4: 375-378, 1984.
175. Cooper, G.M., and Neiman, P.E. Two distinct candidate transforming genes of lymphoid leukosis virus-induced neoplasms. Nature (Lond.), 292: 857-858, 1981.
176. Sprent, J. Migration and lifespan of lymphocytes. In: F. Loor and G.E. Roelants (eds.), Band I cells in immune recognition, pp. 59-82. New York: John Wiley Sons, 1977.
177. Hayward, W.S. Multiple mechanisms of oncogene activation in viral and nonviral neoplasia. In: I.L. Weissman (ed.), Leukemia, Dahlem Konferenzen Berlin, pp. 147-162. Berlin: Springer-Verlag, 1985.
178. Dofuku, R., Biedler, J.L., Spengler, B.A., and Old, L.J. Trisomy of chromosome 15 in spontaneous leukemia of AKR mice. Proc. Nat'l. Acad. Sci. USA, 72: 1515-1517, 1975.
179. Wiener, F., Ohno, S., Spira, J., Haran-Ghera, N., and Klein, G. Chromosome changes (trisomies, no. 15 and 17) associated with tumor progression in leukemias induced by radiation leukemia virus. J. Nat'l. Cancer Inst., 61: 227-237, 1978.
180. Spira, J., Wiener, F., Ohno, S., and Klein, G. Is MLV or DMBA trisomy cause or consequence of murine T-cell leukemia development? Studies on Robertsonian translocation in mice. Proc. Nat'l. Acad. Sci. USA, 76: 6619-6621, 1979.
181. Dofuku, R., Utakoij, T., and Matsuzawa, A. Trisomy of chromosome 13 in spontaneous mammary tumors of GR, C3H and non-inbred Swiss mice. J. Nat'l. Cancer Inst., 63: 651-656, 1979.
182. Hare, W.C.C., Yang, T.-J., and McFreely, R.A. A survey of chromosome findings in 47 cases of bovine lymphosarcoma (leukemia). J. Nat'l. Cancer Inst., 38: 383-392, 1967.
183. Sadamori, N., Kusano, M., Nishimo, K., Fagawa, M., Yao, E., Yamada, Y., Amagasaki, T., Kinoshita, K., and Ichimara, M. Abnormalities of chromosome 14 at band 14g11 in Japanese patients with adult T-cell leukemia. Cancer Genet. Cytogenet., 17: 279-282, 1985.
184. Rowley, J.D., Haven, J.M., Wong-Staal, F., Franchini, G., Gallo, R., and Blattner, W. Chromosome pattern in cells from patients positive for human T-cell leukemia/lymphoma virus. In: R.C. Gallo, M. Essex, and L. Gross (eds.), Human T-cell Leukemia/Lymphoma Virus, pp. 85-89. Cold Spring Harbor, NY: Cold Spring Harbor Press, 1984.
185. Sanada, I., Tanaka, R., Kumagai, E., Tsuda, H., Nishimura, H., Yamaguchi, K., Kawano, F., Fujiwara, H., and Takatsuki, K. Chromosomal aberrations in adult T cell leukemia: relationship to the clinical severity. Blood 65: 649-654, 1985.
186. Pandolfi, F. T-CLL and allied diseases: new insights into classification and pathogenesis. Dign. Immunol., 4: 61-74, 1986.
187. Pandolfi, F., De Rossi, G., and Semenzato, G. T-helper phenotype leukemias: role of HTLV-I. Lancet, 2: 1367-1368, 1985.
188. Fialkow, P.J., and Singer, J.W. Tracing development and cell lineages in human hemopoietic neoplasia. In: I.L. Weissman (ed.), Leukemia, Dahlem Konferenzen Berlin, pp. 203-222. Berlin: Springer-Verlag, 1985.
189. Ueshima, Y., Rowley, J.D., Variakojis, D., Winter, J., and Gordon, L. C
198. Haran-Ghera, N. Pathogenesis of murine leukemia. In: G. Klein (ed.), Viral Oncology, pp. 161-185. New York: Raven Press, 1980.
199. Chang, T.D., Biedler, J.L., Stockert, E., and Old, L.J. Trisomy of chromosome 15 in X-ray-induced mouse leukemia. Proc. Soc. Am. Assoc. Cancer Res., 18: 225, 1977.
200. Klein, G. Lymphoma development in mice and humans: diversity of initiation is followed by convergent cytogenetic evolution. Proc. Nat'l. Acad. Sci. USA, 76: 2442-2446, 1979.
201. Spira, J., Babowits, M., Wiener, F., Ohno, S., Wirschubski, Z., Haran-Ghera, N., and Klein, G. Non-random chromosomal changes in Thy-1-positive and Thy-1 negative lymphomas induced by 7, 12-dimethylbenzanthracene in S26 mice. Cancer Res., 40: 2609-2616, 1980.
202. Erikson, J., Williams, D.L., Finan, J., Howell, P.C., and Croce, C. Locus of the (a)-chain of the T-cell receptor is split by chromosome translocation in T-cell leukemias. Science (Wash. DC), 229: 784-786, 1985.
203. Hecht, F., Morgan, R., Kaiser-McCaw-Hecht, B., and Smith, S. Common region on chromosome 14 in T-cell leukemia and lymphoma. Science (Wash. DC), 226: 1445-1447, 1984.
204. Chen, I.S.Y., Slamon, D.J., Rosenblatt, J.D., Shah, N.P., Quan, S.G., and Wachsman, W. The x gene is essential for HTLV replication. Science (Wash. DC), 229: 54-58, 1985.
205. Haas, M., Mally, M.I., Bogenberger, J.M., Bogart, M.H., Buchannan, M.A., Augery-Bourget, Y., Hyman, R., and Vogt, M. Autocrine growth and progression of murine X-ray-induced T cell lymphomas. EMBO J., 5: 1775-1782, 1986.
206. Oliff, A., McKinney, M.D., and Agranovsky, O. Contribution of the gag and pol sequences to the leukemogenicity of Friend murine leukemia virus. J. Virol., 54: 864-868, 1985.
207. Gazzolo, L., Moscovici, C., and Moscovici, M.G. Persistence of avian oncoviruses in chicken macrophages. Infect. Immun., 23: 294-297, 1979.
208. Schwarz, H., Ihle, J.N., Wecker, E., Fischinger, P.O., Thiel, H.-J., Bolognesi, D.P., and Schafer, W. Properties of mouse leukemia viruses. XVI. Factors required for successful treatment of spontaneous AKR leukemia by antibodies against gp71. Virology, 111: 568-578, 1981.
209. Marx, J.L. AIDS virus has new name-perhaps. Science (Wash. DC), 232: 699-700, 1986.
210. Levy, J.A., Hoffman, A.D., Kramer, S.M., Landis, J.A., Shimabukuro, J.M., and Oshiro, L.S. Isolation of lympho-cytopathic retroviruses from San Francisco patients with AIDS. Science (Wash. DC), 225: 840-842, 1984.
211. Gallo, R.C., Salahuddin, S.Z., Popovic, M., Shearer, G.M., Kaplan, M., Haynes, B.F., Palher, F.J., Redfield, R., Oleske, J., Safai, B., White, F., Foster, P., and Markham, P.D. Frequent detection and isolation of cytopathic retroviruses (HTLV III) from patients with AIDS and at risk for AIDS. Science (Wash. DC), 224: 500-503, 1984.
212. Salahuddin, S.A., Markham, P.D., Popovic, M., Sarngadharan, M.G., Orndorff, S., Fladagar, A., Patel, A., Gold, J., and Gallo, R.C. Isolation of infectious T-cell leukemia/lymphoma virus type III (HTLV-III) from patients with acquired immuno-deficiency syndrome (AIDS) or AIDS-related complex (ARC) and from healthy carriers: a study of risk groups and tissue sources. Proc. Nat'l. Acad. Sci. USA, 82: 5530-5534, 1985.
213. Levy, J.A., Kaminsky, L.S., Morrow, M.J.W., Steiner, K., Luciw, P., Dina, D., Hoxie, J., and Oshiro, L. Infection by the retrovirus associated with the acquired immunodeficiency syndrome. Ann. Intern. Med., 103: 694-699, 1985.
214. Levy, J.A., and Shimabukuro, J. Recovery of AIDS-associated retroviruses from patients with AIDS or AIDS-related conditions and from clinically healthy individuals. J. Infect. Dis., 152: 734-738, 1985.
215. Gottlieb, M.S., Schroff, R., Schamber, H.M., Weisman, J.D., Fan, P.T., Wolf, R.A., and Saxon, A. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immuno-deficiency. N. Engl. J. Med., 305: 1425-1431, 1981.
216. Centers for Disease Control. Weekly surveillance report, October 20, 1986.
217. Sarngadharan, M.G., Popovic, M., Brach, L., Schupbach, J., and Gallo, R.C. Antibodies reactive with human T-lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science (Wash. DC), 224: 506-508, 1984.
218. Blattner, W.A., Biggar, R.J., Weiss, S.H., Melbye, M., and Goedert, J.J. Epidemiology of human T-lymphotropic virus type III and the risk of the acquired immunodeficiency syndrome. Ann. Intern. Med., 103: 665-670, 1985.
219. Kaminsky, L.S., McHugh, T., Stites, D., Volberding, P., Henle, G., Henle, W., and Levy, J.A. High prevalence of antibodies to acquired immune deficiency syndrome (AIDS)-associated retrovirus (ARV) in AIDS and related conditions but not in other disease states. Proc. Nat'l. Acad. Sci. USA, 82: 5535-5539, 1985.
220. Peterman, T.A., Jaffe, H.W., Feorino, P.M., Haverkos, H.W., Stoweburner, R.L., and Curran, J.W. Transfusion-associated acquired immunodeficiency syndrome in the United States. JAMA, 254: 2913-2917, 1985.
221. Clumeck, N., Robert-Guroff, M., Van De Perre, P., Jennings, A., Sibomana, J., Demol, P., Cran, S., and Gallo, R.C. Sero-epidemiological studies of HTLV-III antibody prevalence among selected groups of heterosexual Africans. JAMA, 254: 2599-2602, 1985.
222. Gottlieb, M.S., Wolfe, P.R., Fahey, J.L., Knight, S., Hardy, D., Eppolito, L., Ashida, E., Patel, A., Beall, G.N., and Sun, N. The syndrome of persistent generalized lymphodenopathy: experience with 101 patients. Adv. Exp. Med. Biol., 187: 85-91, 1985.
223. Curran, J.W., Morgan, M.W., Hardy, A.M., Jaffe, H.W., Darrow, W.W., and Dowdle, W.R. The epidemiology of AIDS: current status and future prospects. Science (Wash. DC), 229: 1352-1357, 1985.
224. Sivak, S.L., and Wormser, G.P. How common is HTLV-III infection in the United States? N. Engl. J. Med., 313: 1352, 1985.
225. McDougal, J.S., Cort, S.P., Kennedy, M.S., Cabridilla, C.D., Feorino, P.M., Francis, D.P., Hicks, D., Kalyanaraman, V.S., and Martin, L.S. Immunoassay for the detection and quantitation of infectious human retrovirus, lymphadenopathy-associated virus (LAV). J. Immunol. Methods, 76: 171-183, 1985.
226. Robert-Guroff, M., Brown, M., and Gallo, R.C. HTLV-III neutralizing antibodies in patients with AIDS and ARC. Nature (Lond.), 316: 72-74, 1985.
227. Weiss, R.A., Clapham, P.R., Cheingsong-Popov, R., Dalgleish, A.G., Carne, C.A., Weller, I.V.D., and Tedder, R.S. Neutralization of human T-lymphotropic virus type III by sera of AIDS and AIDS-risk patients. Nature (Lond.), 316: 69-72, 1985.
228. Saxinger, C., Levine, P.H., Lange-Wantzin, G., and Gallo, R. Unique pattern of HTLV-III (AIDS-related) antigen recognition by sera from African children in Uganda (1972). Cancer Res. (Suppl.), 45: 4624-4626, 1985.
229. Bayley, A.C., Cheingsong, Popov, R., Dalgleish, A.G., Downing, R.G., Tedder, R.S., and Weiss, R.A. HTLV-III serology distinguishes atypical and endemic Kaposi's sarcoma in Africa. Lancet, 1: 359-361, 1985.
230. Black, P.H. HTLV-III, AIDS, and the brain. N. Engl. J. Med., 313: 1538-1540, 1985.
231. Resnick, L., DiMarzo-Veronese, F., Schupbach, J., Tortellotte, W.W., Ho, D.D., Muller, F., Shapshak, P., Vogt, M., Gropman, J.E., Markham, P.D., and Gallo, R.C. Intra-blood-brain-barrier synthesis of HTLV-III-specific IgG in patients with neurologic symptoms associated with AIDS or AIDS-related complex. N. Engl. J. Med., 313: 1498-1510, 1985.
232. Ho, D.D., Rota, T.R., Schooley, R.T., Kaplan, J.C., Allan, J.D., Groopman, J.E., Resnick, L., Felsenstein, D., Andres, G.A., and Hirsch, M.S. Isolation of HTLV-III from cerebrospinal fluid and neural tissue of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N. Engl. J. Med., 313: 1493-1497, 1985.
233. Goedert, J.J., Biggar, R.J., Weiss, S.H., Eyster, M.E., Melbye, M., Wilson, S., Ginzburg, H.M., Grossman, R.J., DiGiola, R.A., Sanchez, W.C., Giron, J.A., Ebbesen, P., Gallo, R.C., and Blattner, W.A. Three-year incidence of AIDS in five cohorts of HTLV-III-infected risk group members. Science (Wash. DC), 231: 992-995, 1986.
234. Biggar, R.J. The AIDS problem in Africa. Lancet, 1: 79-83, 1986.
235. Kreiss, J.K., Koech, D., Plummer, F.A., Holmes, K.K., Lightfoote, M., Piot, P., Ronald, A.R., Ndinya-Achola, J.O., D'Costa, L.J., Roberts, P., Ngugi, E.N., and Quinn, T.C. AIDS virus infection in Nairobi prostitutes-spread of the epidemic to East Africa. N. Engl. J. Med., 314: 414-417, 1986.
236. Rodriguez, L., Sinangil, F., Godoy, G., Dewhurst, S., Merino, F., and Volsky, D.J. Antibodies to HTLV-III/LAV among aboriginal Amazonian Indians in Venezuela. Lancet, 2: 1098-1100, 1985.
237. Levy, J.A., and Ziegler, J.L. Acquired immune deficiency syndrome is an opportunistic infection and Kaposi's sarcoma results from secondary stimulation. Lancet, 2: 78-81, 1983.
238. Hoxie, J.A., Haggarty, B.S., Rachowski, J.L., Pilsbury, N., and Levy, J.A. Persistent noncytopathic infection of human lymphocytes with AIDS-associated retrovirus. Science (Wash. DC), 229: 1400-1402, 1985.
239. Anonymous Report. Needle-stick transmission of HTLV-III from a patient infected in Africa. Lancet, 2: 1376-1377, 1984.
240. Cooper, D.A., Maclean, P., Finlayson, R., Michelmore, H.M., Gold, J., Donovan, B., Barnes, T.G., Brooke, P., and Penny, R. Acute AIDS retrovirus infection. Definition of a clinical illness associated with seroconversion. Lancet, 1: 537-540, 1985.
241. Eyster, M.E., Goedert, J.J., Sarngadharan, M.G., Weiss, S.H., Gallo, R.C., and Blattner, W.A. Development and early natural history of HTLV-III antibodies in persons with hemophilia. JAMA, 253: 2219-2223, 1985.
242. Feorino, P.M., Jaffe, H.W., Palmer, E., Peterman, T.A., Francis, D.E., Kalzunaran, V.S., Weinstein, R.A., Stoneburner, R.L., Alexander, W.J., Ruersky, C., Getchell, J.P., Weinfeld, D., Haverkos, H.W., Kilbourne, B.W., Nicholson, J.K.A., and Curran, J.W. Transfusion-asociated acquired immune deficiency syndrome. N. Engl. J. Med., 312: 1293-1296, 1985.
243. Sarin, P.S., and Gallo, R.C. The involvement of human T-lymphotropic viruses in T-cell leukemia and immune deficiency. Cancer Rev., 1: p. 1-17, Copenhagen: Einar Munksgaard, 1986.
244. Sande, M.A. Transmission of AIDS. The case against casual contagion. N. Engl. J. Med., 314: 380-382, 1986.
245. Dayton, A.I., Sodroski, J.G., Rosen, G.A., Goh, W.C., and Haseltine, W.A. The trans-activator gene of the human T cell lymphotropic virus type III is required for replication. Cell, 44: 941-947, 1986.
246. Shaw, G.M., Hahn, B.H., Arya, S.K., Groopman, J.E., Gallo, R.C., and Wong-Staal, F. Molecular characterization of human T-cell leukemia (lymphotropic) virus type III in the acquired immune deficiency syndrome. Science (Wash. DC), 226: 1165-1167, 1984.
247. Harper, M.E., Marselle, L.M., Gallo, R.C., and Wong-Staal, F. Detection of lymphocytes expressing T-lymphotropic virus type III in lymph nodes and peripheral blood from infected individuals by in situ hybridization. Proc. Nat'l. Acad. Sci. USA, 83: 772-776, 1986.
248. Folks, T., Powell, D.M., Lightfoote, M.M., Benn, S., Martin, M., and Fauci, A. Induction of HTLV-III/LAV from a nonvirus-producing T-cell line: implications for latency. Science (Wash. DC), 231: 600-602, 1986.
249. Centers for Disease Control. Update: acquired immuno-deficiency syndrome -- United States. Morbidity Mortality Weekly Rep., 34: 245-248, 1985.
250. Beard, J.W. Avian virus growth and their etiological agents. Adv. Cancer Res., 7: 1-127, 1963.
251. Fischinger, P.J., Ihle, J.N., Levy, J.-P., Bolognesi, D.P., Elder, J., and Schaefer, W. Recombinant murine leukemia viruses and protective factors in leukemogenesis. Cold Spring Harbor Conf. Cell Proliferation, 7: 989-1103, 1980.
252. Centers for Disease Control. Update: evaluation of human T-lymphotropic virus type III/lymphadenopathy-associated virus infection in health care personnel-United States. Morbidity Mortality Weekly Rep., 34: 575-578, 1985.
253. Friedland, G.H., Satzman, B.R., Rogers, M.F., Kahl, P.A., Lesser, M.L., Mayers, M.M., and Klein, R.S. Lack of transmission of HTLV-III/LAV infection to household contacts of patients with AIDS or AIDS-related complex with oral candidiasis. N. Engl. J. Med., 314: 344-349, 1986.
254. Klatzmann, D., Barre-Sinoussi, F., Nugeyre, M.T., Dauguet, C., Vilmer, E., Griscelli, C., Brun-Vezinet, F., Rouzioux, C., Gluckman, J.C., Chermann, J.C., and Montagnier, L. Selective tropism of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science (Wash. DC), 225: 59-63, 1984.
255. Barnes, D.M. AIDS-related brain damage unexplained. Science (Wash. DC), 232: 1091-1093, 1986.
256. Lifson, G.D., Feinberg, M.B., Reyes, G.R., Rabin, L., Banapour, B., Chakrabarti, S., Moss, B., Wong-Staal, F., Steiner, K.S., and Engleman, E.G. Induction of CD4-dependent cell fusion by HTLV-III/LAV envelope glycoprotein. Nature (Lond.), 323: 725-728, 1986.
257. Gartner, S., Markovits, P., Markovitz, D., Kaplan, M., Gallo, R., Popovic, M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science (Wash. DC), 233: 215-219, 1986.
258. Stewart, S.J., Fujimoto, J., and Levy, R. Human T lymphocytes and monocytes bear the same leu-3 (T4) antigen. J. Immunol., 136: 3773-3778, 1986.
259. Montagnier, L., Gruest, J., Chamaret, S., Dauguet, C., Axler, C., Guetard, D., Nugeyre, M.T., Barre-Sinoussi, F., Chermann, J.-C., Burnet, J.B., Klatzmann, D., and Gluckman, J.C. Adaptation of lymphodenopathy associated virus (LAV) to replication in EBT-transformed B lymphoblastoid cells lines. Science (Wash. DC), 225: 63-66, 1984.
260. Levy, J.A., Cheng-Mayer, C., Dina, D., and Luciw, P. AIDS retrovirus (ARV) replicates in human and animal fibroblasts transfected with a molecular clone. Science (Wash. DC), 232: 998-1011, 1986.
261. Shaw, G.M., Harper, M.E., Hahn, B.H., Epstein, L.G., Gajdusek, D.C., Price, R.W., Naria, B.A., Petito, C.K., O'Hara, C.J., Groopman, J.E., Cho, E.-S., Oleske, J.M., Wong-Staal, F., and Gallo, R.C. HTLV III infection in brains of children and adults with AIDS encephalopathy. Science (Wash. DC), 277: 177-181, 1985.
262. Daniel, M.D., King, N.W., Letvin, N.L., Hunt, R.D., Seghal, P.K., and Desrosiers, R.C. A new type D retrovirus isolated from macaques with immune-deficiency syndrome. Science (Wash. DC), 223: 602-605, 1984.
263. Letwin, N.L., Daniel, M.A., Seghal, P.K., Chaliloux, L.V., King, N.W., Hunt, R.D., Aldrich, W.R., Holley, K., Schmidt, D.K., and Desrosiersm, R.C. Experimental infection of rhesus monkeys with type D retrovirus. J. Virol., 52: 683-686, 1984.
264. Kanki, P.J., Alroy, J., and Essex, M. Isolation of T-lymphotropic retrovirus related to HTLV-III/LAV from wild-caught African green monkeys. Science (Wash. DC), 230: 951-954, 1985.
265. Fultz, P.N., McClure, H.M., Swenson, R.B., McGrath, C.R., Brodie, A., Getchell, J.P., Jensen, F.C., Anderson, D.C., Broderson, J.R., and Francis, D.P. Persistent infection of chimpanzees with human T-lymphotropic virus type III/lymphodenopathy-associated virus: a potential model for acquired immunodeficiency syndrome. J. Virol., 58: 116-124, 1986.
266. Purtilo, D.T., Kipscomb, H., Krueger, G., Sonnabend, J., Casareale, D., and Volsky, D.J. Role of Epstein-Barr virus in acquired immune deficiency syndrome. Adv. Exp. Med. Biol., 187: 53-65, 1985.
267. Peutherer, J.F., Edmond, E., Simmonds, P., Dickson, J.D., and Bath, G.E. HTLV-III antibody in Edinburgh drug addicts. Lancet, 2: 1129-1130, 1985.
268. Koprowski, H., DeFreitas, E., Harper, M.E., Sandberg-Wollheim, M., Sheremata, W., Robert-Guroff, M., Saxinger, C.W., Feinberg, M.B., Wong-Staal, F., and Gallo, R.C. Multiple sclerosis and human T-cell lymphoytropic retroviruses. Nature, 318: 154-160, 1985.
269. Lairmore, M.D., Rosadio, R.H., and De Martini, J.C. Ovine lentivirus lymphoid interstitial pneumonia. Rapid induction in neonatal lambs. Am. J. Path., 125: 173-181, 1986.
270. Chen, I.Y., Wachsman, W., Rosenblatt, J.D., and Cann, A.J. The role of the X gene in HTLV associated malignancy. Cancer Surveys 5 (2): 331-342, 1986.
271. Gallo, R.C. The first human retrovirus. Scientific American, 255 (6): 88-98, 1986.
272. Zijlstra, M., and Melief, C.J.M. Virology, genetics and immunology of murine lymphomagenesis. Biochem. et. Biophys. Acta, 865: 197-231, 1986.
273. Duesberg, P.H. Cancer genes: rare recombinants instead of activated oncogenes. Proc. Nat'l. Acad. Sci. USA, in press, 1987.
274. Stoler, M.H., Eskin, T.A., Been, S., Angerer, R.C., and Angerer, L.M. Human T-cell lymphotropic virus type III infection of the central nervous system. J. Am. Med. Assoc., 256: 2360-2364, 1986.
275. Chayt, K.J., Harper, M.E., Marselle, L.M., Lewin, E.B., Rose, R.M., Oleske, J.M., Epstein, L.G., Wong-Staal, F., and Gallo, R.C. Detection of HTLV-III RNA in lungs of patients with AIDS and pulmonary involvement. J. Am. Med. Assoc., 256: 2356-2371, 1986.
276. Scott, G.B., Fischl, M.A., Klimas, N., Fletcher, M.A., Dickinson, G.M., Levine, R.S., and Parks, W.P. Mothers of infants with the acquired immuno-deficiency syndrome. J. Am. Med. Assoc., 253: 363-366, 1985.
277. Gonda, M., Wong-Staal, F., Gallo, R., Clements, J., Narayan, O., and Gilden, R. Sequence homology and morphologic similarity of HTLV-III and visna virus: a pathogenic lentivirus. Science, 227: 173-177, 1985.
278. Walker, C.M., Moody, D.J., Stites, D.P., and Levy, J.A. CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science, 234: 1563-1566, 1986.